SCANMODE problem
-
Hi all, trying to eliminate a negative frequency via SCANMODE. Job aborts with no particular message before scanning. I have FREQINFO file for the restart
FREQINFO.DAT INPUT.d12 input.f34 input.outEXTERNAL FREQCALC RESTART SCANMODE 1 -10 10 0.4 1 END ENDSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS SCAN ALONG NORMAL MODES STARTING POINT: -10 ENDING POINT: 10 STEP: 0.40000 (THE STEP IS GIVEN AS TIMES OF THE CLASSICAL AMPLITUDE AT THE QUANTUM GROUND STATE ENERGY. THE MAX ATOMIC DISPLACEMENT IN THE STEP IS GIVEN IN BOHR WITHIN SQUARE BRACKETS) MODE(CM-1) DISPLAC TOTAL ENE(DFT)(AU) CLASSICAL HARM ENE(AU) NCYC DE [MAX DISP -NATOM-] 1( -62.1) [ 0.113 - 3-] -
fort.f34 INPUT.d12 job.out Further on this problem - I managed to run this without restart - I am notoriously bad restarting. Tryiong to get rid of this small negative frequenyc. I do not see from the scan any problems, e.g. any local minimum I could restart calculation from. It just decreases monotonically...
MODE(CM-1) DISPLAC TOTAL ENE(DFT)(AU) CLASSICAL HARM ENE(AU) NCYC DE [MAX DISP -NATOM-] 1( -62.1) [ 0.113 - 3-] -4.0000 -0.1610002871486E+04 -0.1610016919330E+04 43 -0.9E-10 -3.6000 -0.1610006913204E+04 -0.1610016489435E+04 32 -0.4E-10 -3.2000 -0.1610009918764E+04 -0.1610016104792E+04 34 0.2E-11 -2.8000 -0.1610012035843E+04 -0.1610015765400E+04 37 -0.8E-11 -2.4000 -0.1610013425826E+04 -0.1610015471261E+04 39 0.2E-08 -2.0000 -0.1610014243529E+04 -0.1610015222375E+04 121 0.4E-09 -1.6000 -0.1610014634134E+04 -0.1610015018740E+04 39 -0.5E-12 -1.2000 -0.1610014749019E+04 -0.1610014860357E+04 78 0.4E-10 -0.8000 -0.1610014729596E+04 -0.1610014747227E+04 54 -0.1E-09 -0.4000 -0.1610014679529E+04 -0.1610014679349E+04 36 0.1E-11 0.0000 -0.1610014656723E+04 -0.1610014656723E+04 CENTRAL POINT -
Dear Jonas,
I took your provided data and plotted it. At first, it indeed seems as monotonically decreasing along one side of the quadratic function, but if you zoom in closely, you will notice this little "drop":
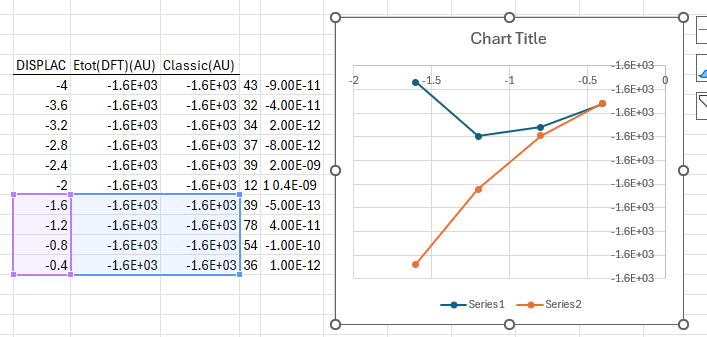 .
.I suggest you do a second scanning along the same vibrational mode, but on a finer grid (e.g., step of 0.05) in the interval -1.6 / +1.6. Then take the geometry from one of the local minima (to the left and to the right of the central point, they need to be equivalent) and optimise it. The subsequent frequencies should be positive, unless there is a second negative mode or your structure is intrinsically unstable at these conditions.
Hope this helps.
Cheers,
Aleks -
No idea... I just have no idea... I ran a complete run and all of a sudden the same abort. So frustrating since I do not understand the error
SSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS SCAN ALONG NORMAL MODES STARTING POINT: -1 ENDING POINT: 0 STEP: 20.00000 (THE STEP IS GIVEN AS TIMES OF THE CLASSICAL AMPLITUDE AT THE QUANTUM GROUND STATE ENERGY. THE MAX ATOMIC DISPLACEMENT IN THE STEP IS GIVEN IN BOHR WITHIN SQUARE BRACKETS) MODE(CM-1) DISPLAC TOTAL ENE(DFT)(AU) CLASSICAL HARM ENE(AU) NCYC DE [MAX DISP -NATOM-] 1( -62.7) [ 5.606 - 3-] Abort(1) on node 31 (rank 31 in comm 0): application called MPI_Abort(MPI_COMM_WORLD, 1) - process 31 -
Oh, I see... somewhere among the files I found this. It is unusual since everything converged optimized/converged otherwise. Now wonder I can't restart, it is not restart problem. Why would i become linearly dependent in SCANMODE but not before?
ERROR **** CHOLSK **** BASIS SET LINEARLY DEPENDENT
-
Dear Jonas,
The basis set linear dependence issue might arise from the fact that the geometry in the scan step changes too much from the starting point and hence the interatomic distance changes a lot. Your scan step seems rather large, maybe that is the underlying issue. You can print out the geometry that will be scanned (by setting the first number as a negative values, i.e., "-1") and check that it is sensible. But perhaps I would first try with the smaller step, say 0.4 as in the first case and see if that helps.
Cheers,
Aleks -
Hi Aleks, I tried but ended up just using LDREMO keyword
Another superstrange thing (that of course was not happening yesterday), that I can't seem to specify initial and final step anymore properly, it divides it by 10... Bizzare. I now have to multiply initial step by 10 (-1.6 start I want I need to specify as 16!) so it startes at -1.6...My input is:
EXTERNAL FREQCALC RESTART SCANMODE 1 -16 0 0.1 1 END ENDSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS SCAN ALONG NORMAL MODES STARTING POINT: -16 ENDING POINT: 0 STEP: 0.10000 (THE STEP IS GIVEN AS TIMES OF THE CLASSICAL AMPLITUDE AT THE QUANTUM GROUND STATE ENERGY. THE MAX ATOMIC DISPLACEMENT IN THE STEP IS GIVEN IN BOHR WITHIN SQUARE BRACKETS) MODE(CM-1) DISPLAC TOTAL ENE(DFT)(AU) CLASSICAL HARM ENE(AU) NCYC DE [MAX DISP -NATOM-] 1( -62.7) [ 0.028 - 3-] -1.6000 -0.1610014638959E+04 -0.1610015023766E+04 18 -0.1E-07 -1.5000 -0.1610014685887E+04 -0.1610014979469E+04 29 0.5E-06 -1.4000 -0.1610014719434E+04 -0.1610014938029E+04 13 -0.9E-07 -1.3000 -0.1610014740251E+04 -0.1610014899447E+04 13 -0.2E-06 -1.2000 -0.1610014750671E+04 -0.1610014863723E+04 21 -0.9E-08 -1.1000 -0.1610014754239E+04 -0.1610014830856E+04 15 -0.4E-06 -1.0000 -0.1610014749993E+04 -0.1610014800848E+04 28 0.1E-07 -0.9000 -0.1610014740870E+04 -0.1610014773698E+04 23 0.1E-05 -
Either way, seems you got your geometry for further optimization
")
Cheers,
A