Filippo Bodo
Posts
-
BASISSET mTZVP not allowed -
CLSEGV **** NOT PAIRING AT K -
CLSEGV **** NOT PAIRING AT KHi Alexander,
I took a look into your calculations and I performed a few tests on them.As you said, the calculation seems to break at the end of the optimization as it enters in the
FINALRUN. This is because when you perform a geometry optimization the bielectronic integrals are indexed based on the starting geometry and they are kept the same throughout the entire optimization process to avoid any numerical instabilities. WithFINALRUNactive, the code performs a new indexing that in your case leads to the issue you observed.Working a bit with the
OPTGEOMinput you gave us I found two possible workarounds:- You can either deactivate the
FINALRUNoption, and if you feel safer you can perform a second optimization on the geometry you obtain
[...] OPTGEOM FINALRUN 0 END [...]- An alternative option is instead to increase the
TOLINTEGused in the optimization, and, in this case, I didn't encounter any issue using
[...] TOLINTEG 10 10 10 10 20 [...]For both cases I have attached an output below
opt_finalrun.out
opt_tolinteg.out - You can either deactivate the
-
Electronic Structure Bugs in CRYSTALClear -
pob-TZVP-rev2 library errorDear Alexander,
We runned a few tests, and, indeed, we found the same behavior. This, though, is not due to an error in the definition in the basis set, but rather to some formatting issue, since the Cs goes up to P4 the code expects that also the Iodine pseudo goes up to P4 and fills the missing coefficients and exponents with zeros.Luckily there is an easy workaround to this, it is sufficient to flip Cs and I definition in the geometry and the results are the same as the one you obtained by defining the basis set in the input as you can see from my test.
I will leave you here two output snippets hoping that they help clarifying the issue:
- Cs defined before I in the geometry section
INPUT COORDINATES ATOM AT. N. COORDINATES 1 38 5.000000000000E-01 5.000000000000E-01 5.000000000000E-01 2 55 0.000000000000E+00 0.000000000000E+00 0.000000000000E+00 3 53 0.000000000000E+00 5.000000000000E-01 5.000000000000E-01 [...] ******************************************************************************* *** PSEUDOPOTENTIAL INFORMATION *** ******************************************************************************* ATOMIC NUMBER 38, NUCLEAR CHARGE 10.000, PSEUDOPOTENTIAL TYPE EXPONENT COEFF. N EXPONENT COEFF. N P0 TMS 6.9334610 135.2710429 0 4.1140038 17.9440714 0 P1 TMS 7.2168166 29.4380813 0 7.1736962 58.8806749 0 3.0227988 4.9362827 0 2.8656990 9.7233521 0 P2 TMS 6.3215146 11.9072392 0 6.3914995 17.8595514 0 1.7697266 2.1991802 0 1.6367717 2.8935709 0 P3 TMS 4.2441984 -5.5093333 0 4.2291645 -7.3046417 0 ATOMIC NUMBER 55, NUCLEAR CHARGE 9.000, PSEUDOPOTENTIAL TYPE EXPONENT COEFF. N EXPONENT COEFF. N P0 TMS 4.0811192 84.5477223 0 2.4215224 16.6540350 0 P1 TMS 5.5339726 52.3496307 0 5.5067944 104.6994132 0 2.2809616 8.8065577 0 2.1034905 17.6166111 0 P2 TMS 1.8131494 5.2689855 0 1.8077217 7.9036419 0 0.8729040 1.3364313 0 0.8587203 2.0056513 0 P3 TMS 5.2170839 -16.4976543 0 5.1481965 -23.3081313 0 1.5805995 -2.2368273 0 1.3478959 -2.2269420 0 P4 TMS 1.8077398 -2.5041987 0 1.8050613 -3.1382445 0 ATOMIC NUMBER 53, NUCLEAR CHARGE 25.000, PSEUDOPOTENTIAL TYPE EXPONENT COEFF. N EXPONENT COEFF. N P0 TMS 40.0333760 49.9896490 0 17.3005760 281.0065560 0 8.8517200 61.4167390 0 P1 TMS 15.7201410 67.4162390 0 15.2082220 134.8076960 0 8.2941860 14.5665480 0 7.7539490 28.9684220 0 P2 TMS 13.8177510 35.5387560 0 13.5878050 53.3397590 0 6.9476300 9.7164660 0 6.9600990 14.9775000 0 P3 TMS 18.5229500 -20.1766180 0 18.2510350 -26.0880770 0 7.5579010 -0.2204340 0 7.5974040 -0.2216460 0 P4 TMS 0.0000000 0.0000000 0 0.0000000 0.0000000 0- I defined before Cs in the geometry section
INPUT COORDINATES ATOM AT. N. COORDINATES 1 38 5.000000000000E-01 5.000000000000E-01 5.000000000000E-01 2 53 0.000000000000E+00 5.000000000000E-01 5.000000000000E-01 3 55 0.000000000000E+00 0.000000000000E+00 0.000000000000E+00 [...] ******************************************************************************* *** PSEUDOPOTENTIAL INFORMATION *** ******************************************************************************* ATOMIC NUMBER 38, NUCLEAR CHARGE 10.000, PSEUDOPOTENTIAL TYPE EXPONENT COEFF. N EXPONENT COEFF. N P0 TMS 6.9334610 135.2710429 0 4.1140038 17.9440714 0 P1 TMS 7.2168166 29.4380813 0 7.1736962 58.8806749 0 3.0227988 4.9362827 0 2.8656990 9.7233521 0 P2 TMS 6.3215146 11.9072392 0 6.3914995 17.8595514 0 1.7697266 2.1991802 0 1.6367717 2.8935709 0 P3 TMS 4.2441984 -5.5093333 0 4.2291645 -7.3046417 0 ATOMIC NUMBER 53, NUCLEAR CHARGE 25.000, PSEUDOPOTENTIAL TYPE EXPONENT COEFF. N EXPONENT COEFF. N P0 TMS 40.0333760 49.9896490 0 17.3005760 281.0065560 0 8.8517200 61.4167390 0 P1 TMS 15.7201410 67.4162390 0 15.2082220 134.8076960 0 8.2941860 14.5665480 0 7.7539490 28.9684220 0 P2 TMS 13.8177510 35.5387560 0 13.5878050 53.3397590 0 6.9476300 9.7164660 0 6.9600990 14.9775000 0 P3 TMS 18.5229500 -20.1766180 0 18.2510350 -26.0880770 0 7.5579010 -0.2204340 0 7.5974040 -0.2216460 0 ATOMIC NUMBER 55, NUCLEAR CHARGE 9.000, PSEUDOPOTENTIAL TYPE EXPONENT COEFF. N EXPONENT COEFF. N P0 TMS 4.0811192 84.5477223 0 2.4215224 16.6540350 0 P1 TMS 5.5339726 52.3496307 0 5.5067944 104.6994132 0 2.2809616 8.8065577 0 2.1034905 17.6166111 0 P2 TMS 1.8131494 5.2689855 0 1.8077217 7.9036419 0 0.8729040 1.3364313 0 0.8587203 2.0056513 0 P3 TMS 5.2170839 -16.4976543 0 5.1481965 -23.3081313 0 1.5805995 -2.2368273 0 1.3478959 -2.2269420 0 P4 TMS 1.8077398 -2.5041987 0 1.8050613 -3.1382445 0I hope this helps
-
Unexpectedly cannot get the geometry after optimization: KEYWORD EXTPRT NOT ALLOWEDDear esmuigors
esmuigors said in Unexpectedly cannot get the geometry after optimization: KEYWORD EXTPRT NOT ALLOWED:Should I only take the atoms labelled by "T" if the lattice is tetragonal, and which ones are to be taken in case of an orthorombic one? Or does "T"'and "F" stand just for "TRUE"'and "FALSE"?
You are correct T and F stand for 'TRUE' and 'FALSE'.
esmuigors said in Unexpectedly cannot get the geometry after optimization: KEYWORD EXTPRT NOT ALLOWED:
I am actually talking about the standard runPcry23 script. Perhaps it should not be like that? Or should I modify this script?
That script already contain the few line of codes I suggested you, if not feel free to modify it and add those lines.
esmuigors said in Unexpectedly cannot get the geometry after optimization: KEYWORD EXTPRT NOT ALLOWED:
Actually, I have now tried using only the "T"-labeled atoms and got this error:
ERROR **** geometry **** FORMAT ERROR IN INPUT DECK
I checked the input on top of the CS2_B1WC.pob_tzvp_rev2_gamma_onlyT.out file it seems like you are defining six atoms in the asymmetric unit, but only two atomic positions are specified in the input.
CRYSTAL 0 0 0 64 6.34350113 5.54788046 9.71085545 6. ! Definitions of the number of atoms in the asymmetric unit 6 0.000000000000e+00 0.000000000000e+00 -5.000000000000e-01 16 2.303037287581e-17 3.119080311720e-01 1.207617820468e-01 ATOMSYMM ...The number of atoms in the asymmetric unit and the position specified should always match.
I hope this helps you,
Best,
-
Unexpectedly cannot get the geometry after optimization: KEYWORD EXTPRT NOT ALLOWEDesmuigors said in Unexpectedly cannot get the geometry after optimization: KEYWORD EXTPRT NOT ALLOWED:
About the manual extraction – do I understand correctly that I should take the primitive and not crystallographic cell parameters?! The input in the .d12 file is in crystallographic cell, isn't it?
Hi esmuigors,
If you want to proceed with the manual extraction of the geometry you should copy the Crystallographic cell lattice parameters required by the space group you are working with (e.g. Monoclinic will require you a, b, c and β, while a cubic only the a lattice vector), the only exception to this are P lattices where the primitive and crystallographic coincide. Accordingly, also the fractionary coordinate you will copy should be the one of the Crystallographic cell, but you should copy only the atoms present in the asymmetric unit of your system identified by a T in the FINAL OPTIMIZED GEOMETRY print in the .out file, I will include here below an example to better clarify.FINAL OPTIMIZED GEOMETRY - DIMENSIONALITY OF THE SYSTEM 3 (NON PERIODIC DIRECTION: LATTICE PARAMETER FORMALLY SET TO 500) ******************************************************************************* LATTICE PARAMETERS (ANGSTROMS AND DEGREES) - BOHR = 0.5291772083 ANGSTROM PRIMITIVE CELL - CENTRING CODE 6/0 VOLUME= 62.890767 - DENSITY 13.512 g/cm^3 A B C ALPHA BETA GAMMA 6.02167869 6.02167869 6.02167869 147.475653 147.475653 46.660012 ******************************************************************************* ATOMS IN THE ASYMMETRIC UNIT 2 - ATOMS IN THE UNIT CELL: 4 ATOM X/A Y/B Z/C ******************************************************************************* 1 T 273 TA -2.420755613827E-03 -2.420755613827E-03 4.983867094700E-20 2 F 273 TA -2.524207556138E-01 2.475792443862E-01 -5.000000000000E-01 3 T 233 AS 4.184207556138E-01 4.184207556138E-01 -2.220446049250E-16 4 F 233 AS 1.684207556138E-01 -3.315792443862E-01 -5.000000000000E-01 TRANSFORMATION MATRIX PRIMITIVE-CRYSTALLOGRAPHIC CELL 0.0000 1.0000 1.0000 1.0000 0.0000 1.0000 1.0000 1.0000 0.0000 ******************************************************************************* CRYSTALLOGRAPHIC CELL (VOLUME= 125.78153367) A B C ALPHA BETA GAMMA 3.37253732 3.37253732 11.05868170 90.000000 90.000000 90.000000 COORDINATES IN THE CRYSTALLOGRAPHIC CELL ATOM X/A Y/B Z/C ******************************************************************************* 1 T 273 TA -1.956455588613E-19 2.296201495291E-19 -2.420755613827E-03 2 F 273 TA -5.000000000000E-01 2.084229456032E-17 -2.524207556138E-01 3 T 233 AS 5.000000000000E-01 5.000000000000E-01 -8.157924438617E-02 4 F 233 AS -5.117515658395E-17 -5.000000000000E-01 -3.315792443862E-01In this example you'll have to extract from the second geometry print (i.e. Crystallographic cell) the a and c lattice parameters, given the Tetragonal lattice, alongside the fractionary coordinate of atoms 1 and 3.
esmuigors said in Unexpectedly cannot get the geometry after optimization: KEYWORD EXTPRT NOT ALLOWED:
Are there any workarounds to persuade the script to actually copy the fort.34 file?
In this regards, I don't have your script, but it should be sufficient to add to your script something along these lines in the section where all the files are copied back to your working folder:
if [ -e fort.34 ] then cp fort.34 $HERE/$INPUTFILE.f34 fiin this case
$HERE=$PWD, while$INPUTFILEis the variable corresponding to your input name.I hope this helps.
-
forrtl: severe (256): unformatted I/O to unit open for formatted transfers, unit 85, file /dev/null -
forrtl: severe (256): unformatted I/O to unit open for formatted transfers, unit 85, file /dev/null -
Introduction and Installation GuideCRYSTALClear is an open source project that provides an easy Python interface with CRYSTAL. The package allows you to quickly extract information from the CRYSTAL output files and to easily generate customizable plots of computed quantities. In particular, the package supports plotting functionalities for:
Band Structures and DOSS Mechanical Properties IR and Raman Spectra 


QTAIM Transport Properties Density Maps 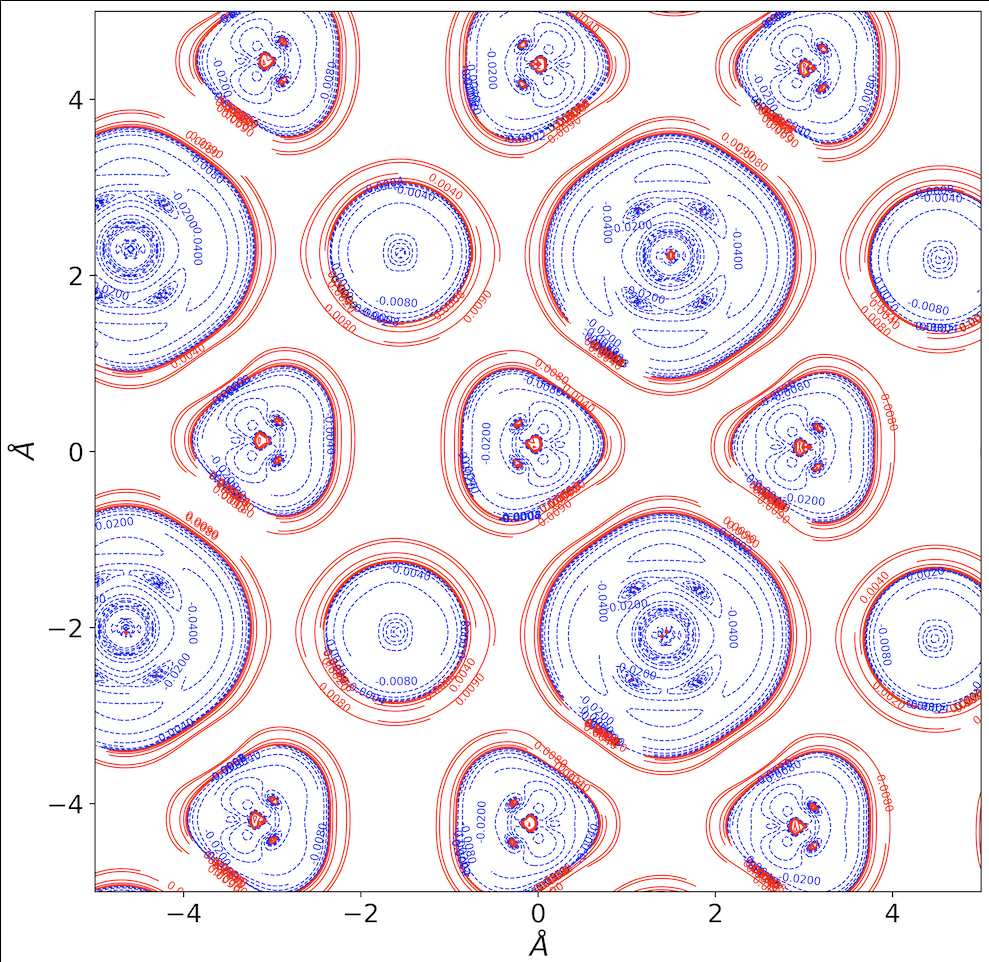
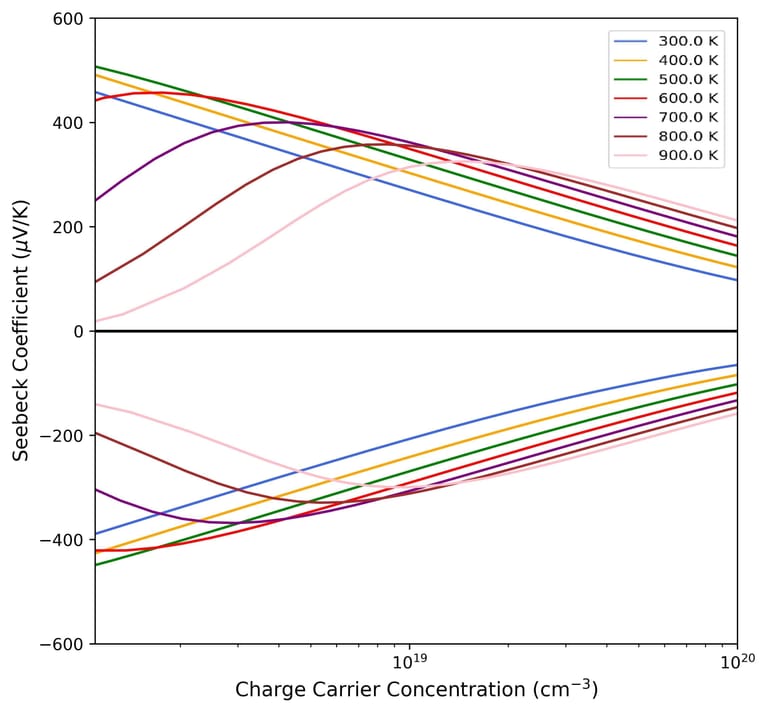

For a more comprehensive review of the package capabilities we refer you to the Documentation Website.
How to Install
Prerequisites:
-
Python: Ensure you have Python installed on your system.
-
conda (Optional but Recommended): conda is a powerful package and environment manager that simplifies the process of managing Python dependencies. If you don't have conda, install it from the official Anaconda distribution: https://www.anaconda.com/
Installation Steps:
- Create a Dedicated Environment (Recommended):
Creating a separate conda environment for CRYSTALClear helps isolate its dependencies from other projects and avoid potential conflicts.
conda create -n env_name python=3.9 # Replace 3.9 with your desired Python version- Activate the Environment (Reccommended):
conda activate env_name- Install CRYSTALClear using pip:
pip install CRYSTALClearThis command will install the CRYSTALClear library and its dependencies.
Verification:
- Check Installation:
After installation, you can verify the installation by importing the library in a Python script or interactive session:
import CRYSTALClearIf the import is successful without any errors, the installation was successful.
Additional Notes:
- Dependencies: The CRYSTALClear library might have dependencies on other libraries. pip will automatically install these dependencies during the installation process.
- Updating: To update the CRYSTALClear library to the latest version:
pip install --upgrade CRYSTALClearTroubleshooting:
- If you encounter any issues during installation:
- Check for any error messages and try to resolve them based on the error descriptions.
- Ensure you have an active internet connection.
- Try updating pip to the latest version:
pip install --upgrade pip - If the issue persists, consider creating a new conda environment and reinstalling the library.
If you the issues persist please feel free to comment here below we will try our best to help you.
How to Use it
In order to use any of the feature available in the package the user need to generate
a CRYSTAL object through thecrystal_iomodule. Three classes are available to the user
for different use cases:- Crystal_output: To extract informations and generate the object required to plot
from any.outfile of the CRYSTAL package. - Properties_output: To generate the objects required to plot any of quantities
stored in the .DAT and .f25 files generated by the PROPERTIES module. - External_unit: To generate the objects required to plot any of the quantites stored
in .DAT and .f25 file generated by CRYSTAL.
Once the object is generated the user can generate the desired plot using the functions
in the plot moduleExample
# Example Band Structure plot from CRYSTALClear.crystal_io import Properties_output import CRYSTALClear.plot as CCplt import matplotlib.pyplot as plt data = Properties_output().read_electron_band() CCplt.plot_electron_band(data, **kwargs) # see the documentation for the available **kwargs # The plotting function returns a matplotlib object that can be visualized as follows plt.show()The plotting function will return one (or a list of) matplotlib object(s) that the user can easily modify as any other plot produced with the library.
For a more comprehensive set of example we refer you to the following jupyter notebook repository.
How to Contribute
Any contribution to the project is more than welcome and greatly appreciated, given the open source nature of the project. If you'd like to do so, please check our guidelines on GitHub
How to Cite
If you use this package in any of your work we kindly ask you to cite the following pubblication:
Camino, Bruno, Huanyu Zhou, Eleonora Ascrizzi, Alberto Boccuni, Filippo Bodo, Alessandro Cossard, Davide Mitoli, Anna Maria Ferrari, Alessandro Erba, and Nicholas M. Harrison, Comput. Phys. Commun. 292, 108853 (2023).
@article{camino2023crystalpytools, title={CRYSTALpytools: A Python infrastructure for the CRYSTAL code}, author={Camino, Bruno and Zhou, Huanyu and Ascrizzi, Eleonora and Boccuni, Alberto and Bodo, Filippo and Cossard, Alessandro and Mitoli, Davide and Ferrari, Anna Maria and Erba, Alessandro and Harrison, Nicholas M}, journal={Computer Physics Communications}, volume={292}, pages={108853}, year={2023}, publisher={Elsevier} } -
