It was my mistake with DOSS. With doss is ok.
QMQDCHEM
@QMQDCHEM
Posts
-
Electronic Structure Bugs in CRYSTALClear -
Electronic Structure Bugs in CRYSTALClear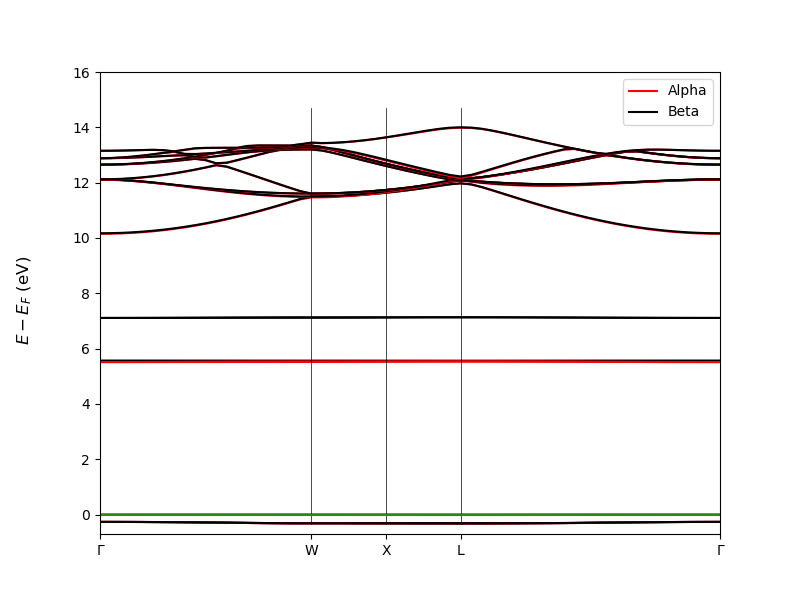
import CRYSTALClear.plot as CCplt import matplotlib.pyplot as plt from pathlib import Path band_files = list(Path('.').rglob('BAND.DAT')) bands = [] for index, band_file in enumerate(band_files): print(f'Processing: {band_file}') band = Properties_output().read_electron_band(str(band_file)) bands.append(band) CCplt.plot_electron_band( band, energy_range=[-0.7, 16], linewidth=1.5, figsize=(8,6), k_labels=['Gamma','W','X','L','Gamma'] ) plt.show() CCplt.plot_electron_band( bands, energy_range=[-1, 16], linewidth=1.5, figsize=(9,7), k_labels=['Gamma','W','X','L','Gamma'] ) plt.show() END_OF_PYTHONI cannot change the height of the lines marking the k-points on the band structure diagram.
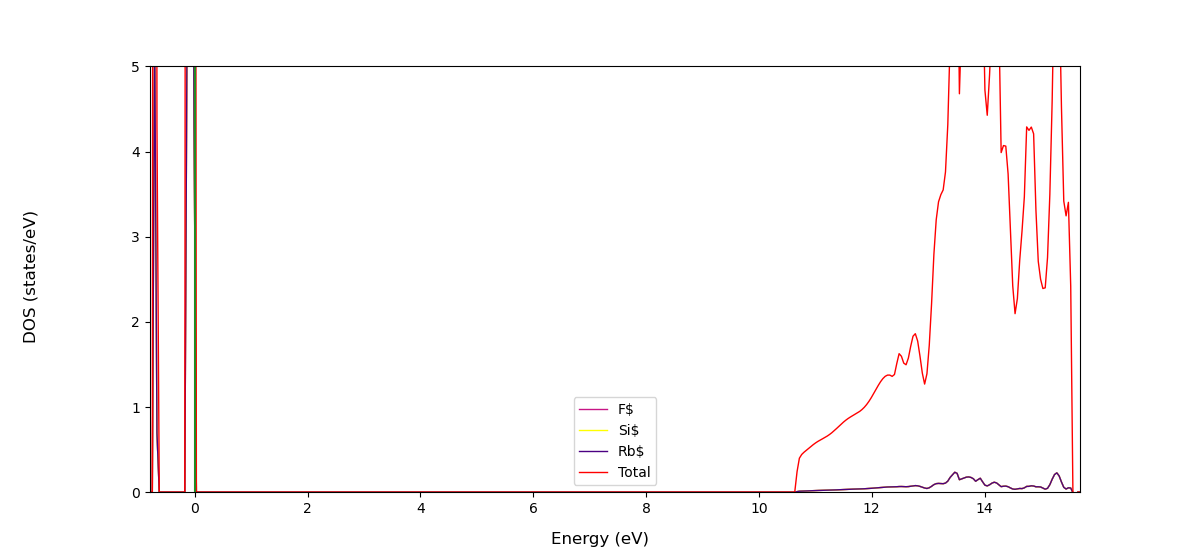
#!/bin/bash source /home/noctua-v2/miniconda3/bin/activate crystal exec python - "$@" <<'END_OF_PYTHON' from CRYSTALClear.crystal_io import Properties_output import CRYSTALClear.plot as CCplt import matplotlib.pyplot as plt # DOSS.DAT path declaration doss = 'DOSS.DAT' # Crystal Object generation doss = Properties_output().read_electron_dos(doss) # Plot generation CCplt.plot_electron_dos(doss, overlap=True, color=['mediumvioletred', 'yellow', 'indigo', 'red'], labels=['F$', 'Si$','Rb$', 'Total'], dos_range=[0,10]) plt.show()I cannot change the y-axis grid on the DOS plot.
-
How use CRYSTALpytools? -
How use CRYSTALpytools?(base) noctua-v2@noctua-v2-X9DR3-F:~/Desktop/RSF/band$ ls BAND.DAT 'BAND STRUCTURE.jpeg' fort.25 fort.87 fort.9 INPUT OUTPUTCan I use BAND.DAT as *.BAND and OUTPUT as *.out files in code?
from CRYSTALpytools.electronics import ElectronBand from pymatgen.electronic_structure.plotter import BSPlotter band = ElectronBand.from_file('*.DAT', 'OUTPUT') pmgband = mgoband.to_pmg(labels=[r'$\Gamma$', 'W', 'X', 'L', r'$\Gamma$']) bsplot = BSPlotter(pmgband) bsplot.get_plot(ylim=(-10, 10), zero_to_efermi=True) -
How create band structure in P1 symmetry -
How compilate CRYSTAL23 on old intell processors? -
How compilate CRYSTAL23 on old intell processors? -
How compilate CRYSTAL23 on old intell processors?First — my apologies. My previous post was written with an automatic translator and was confusing. Sorry for that. Below is a corrected, concise description of the problem and the exact output I have. I would appreciate any advice.
Short summary
- Machine: dual-socket server node with two Intel Xeon E5-2697 v2 (Ivy Bridge). These CPUs support AVX but do not support AVX2.
- Compiler/linker: Intel Fortran (I use mpiifx as the Fortran driver and icc/icpc for C/C++).
- Build: I compiled using the included .inc (snippet below) with -march=ivybridge. Compilation completed successfully.
4)Run: When running the produced binary with the CRYSTAL input (included below) the program crashes immediately with:
forrtl: severe (168): Program Exception - illegal instruction
Image PC Routine Line Source
libc.so.6 0000718B3FC45330 Unknown Unknown Unknown
PcrystalOMP 0000000000E915A2 Unknown Unknown Unknown
PcrystalOMP 0000000006828517 Unknown Unknown Unknown
PcrystalOMP 00000000004073BD Unknown Unknown Unknown
PcrystalOMP 000000000040735D Unknown Unknown Unknown
libc.so.6 0000718B3FC2A1CA Unknown Unknown Unknown
libc.so.6 0000718B3FC2A28B __libc_start_main Unknown Unknown
PcrystalOMP 0000000000407275 Unknown Unknown UnknownMy .inc file:
For Linux based on x86_64 arch, using Intel Fortran Compiler
F90 = mpiifx
LD = $(F90)
PLD = mpiifxF90COMMON = -diag-disable 7373 -diag-error 5198,6182,6893,6916,6919,7374,7416,7423,8089,8586
-align -static-intel -cxxlib -qopenmp
F90FLAGS = $(F90COMMON) -O3 -march=ivybridge
F90BASIS = $(F90COMMON) -O0
F90GORB = $(F90COMMON) -O2
F90DENS = $(F90COMMON) -O2
F90FIXED = -FI
F90FREE = -FR
SAVEMOD = -module $(MODDIR)
INCMOD = -I$(MODDIR)
LDFLAGS = $(F90FLAGS)
EIGENV = $(OBJDIR)/diag_lapack.o
MATMULT = $(OBJDIR)/mult_blas.oMKLPATH = $(MKLROOT)/lib/intel64
LDLIBS = $(LIBXCFUN) -Wl,--start-group
$(MKLPATH)/libmkl_intel_lp64.a $(MKLPATH)/libmkl_intel_thread.a
$(MKLPATH)/libmkl_core.a $(MKLPATH)/libmkl_blacs_openmpi_lp64.a -Wl,--end-group -liomp5 -lpthread -lm -ldl
#LDLIBS =MXMB = $(OBJDIR)/libmxm.o
MACHINE_C=mach_linux
CC = icc
CFLAGS = -O2 -vec-report0 -Wall -diag-disable 177,279,383,869,981,1418,1419,1572 -DNDEBUG
CXX = icpc
CXXFLAGS = $(CFLAGS) -fno-rtti -fno-exceptionsMPI harness
HARNESS = $(MPI)
https://www.intel.com/content/www/us/en/developer/tools/oneapi/onemkl-link-line-advisor.html
MKL=$(MKLROOT)/lib/intel64
MPPLIB=-L$(MKL) $(MKL)/libmkl_scalapack_lp64.a -Wl,--start-group
$(MKL)/libmkl_intel_lp64.a $(MKL)/libmkl_sequential.a
$(MKL)/libmkl_core.a $(MKL)/libmkl_blacs_openmpi_lp64.a -Wl,--end-group -liomp5 -lpthread -lm -ldlThank you in advance for any guidance. I’m happy to attach any of the files or run recommended diagnostics — please tell me which outputs would be most helpful.
Again, sorry for the confusion from the earlier (poorly translated) message.
-
How compilate CRYSTAL23 on old intell processors?I probably won’t be able to share the .inc file at the moment, as I’ve modified it multiple times. The situation is that the compilation completes successfully and crystal23 starts, but it crashes because my processor’s AVX version is too old for the Intel toolkit.
I mentioned the processor model for a reason: https://www.intel.com/content/www/us/en/products/sku/75281/intel-xeon-processor-e52695-v2-30m-cache-2-40-ghz/specifications.html
Support for this CPU was effectively discontinued even before the first versions of the Intel toolkit were released.
I wasn’t able to disable the use of this instruction during compilation. However, if you know how to do it, I can locate one of the working .inc files I still have.
Given this, it seems that using a different compiler would be necessary.
-
How compilate CRYSTAL23 on old intell processors?Hi!
I am trying to compile Crystal 23 on Xeon E-2695v2 processors, but they are not supported by the Intel Toolkit. I am missing the instruction vfmadd231sd, giving GDB output:
Program received signal SIGILL, Illegal instruction.
0x0000000000e915a2 in timer_module_mp_init_timer_ ()
(gdb) bt full
#0 0x0000000000e915a2 in timer_module_mp_init_timer_ ()
No symbol table info available.
#1 0x0000000006828517 in initpp_ ()
No symbol table info available.
#2 0x00000000004073bd in MAIN__ ()
No symbol table info available.
(gdb) info registers
Missing register name
(gdb) x/i $rip
=> 0xe915a2 <timer_module_mp_init_timer_+82>:
vfmadd231sd 0x73c02ed(%rip),%xmm0,%xmm3 # 0x8251898Could you please suggest a compilation method, such as using GCC or any other compiler? Is there a way to compile it using CMake?
Thank you!