Hi Prof. Erba,
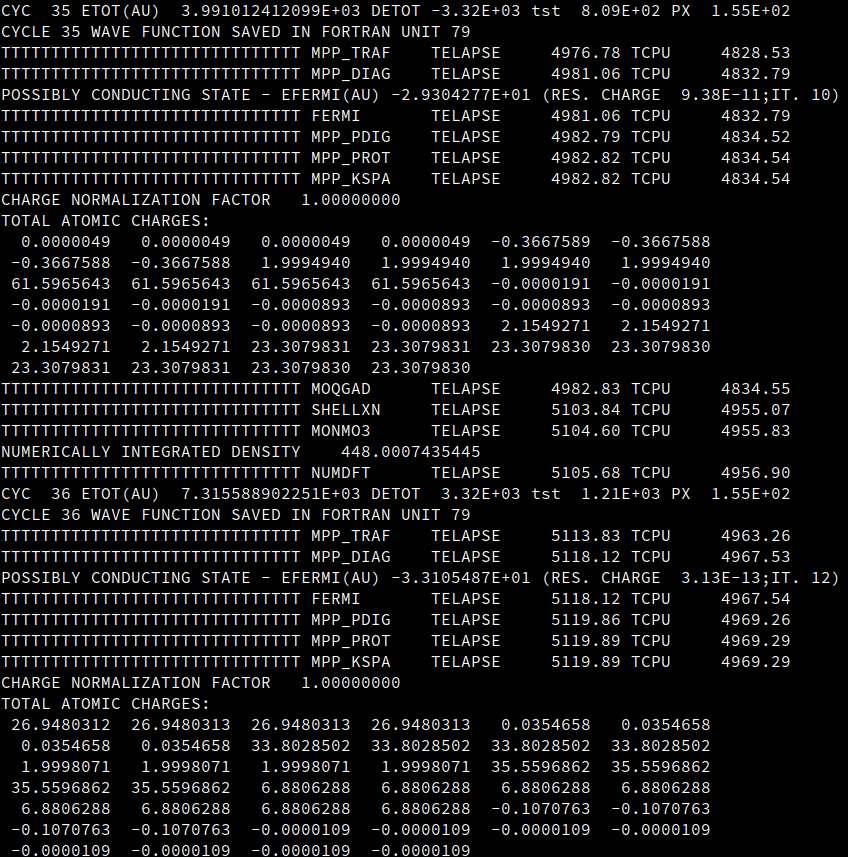
Thanks for the reply. From the file, I could see there are value from energy " -7.9324E-01" (Line No: 81826). I could plot the data using Python, Xmgrace. With CRYSPLOT somehow the data loading is slow and my browser hangs so I am not able to check it. But definitely there is data.
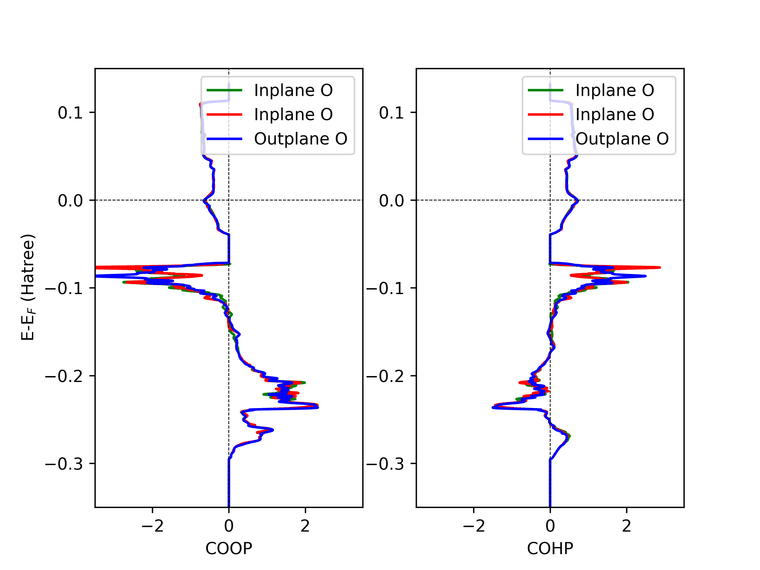
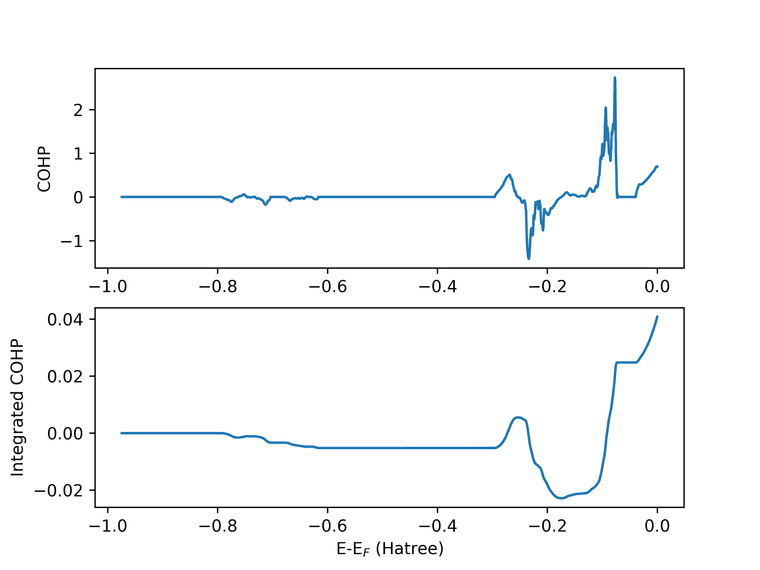
I requested for COHP between Ni and three nearby oxygen. In the file there are 5 columns, so if I am not mistaken it corresponds to "Energy - Next three columns in the order I request - Final Column of DOS ?" (Because the last column data when plotted matches the DOS)
Thanks & Regards,
Rams