P 21/a symmetry not found
-
I am trying to input the following cif file Mg, Acta Crystallographica 17 (1964) 1478-1479.cif which is P 21/a space group 14. However, in the manual space group 14 is P 21/c and my geometry ends up looking funky. I also tried entering manually, e.g. P 2 1a but that simply does not work. Can you help me enter the correct space group here?
CRYSTAL
1 0 0
P 2 1a
9.383 12.669 6.220 107.05
20
7 0.1321 0.3509 0.3611
1 0.058 0.337 0.225
1 0.208 0.305 0.394
1 0.095 0.344 0.487
1 0.174 0.421 0.346
12 0 0 0
8 0.1603 -0.1094 -0.0307
8 0.1685 0.1042 0.1656
8 -0.0017 -0.0687 0.2986
1 0.2 0.091 0.317
1 0.227 0.134 0.116
1 0.252 -0.096 0.059
1 0.143 -0.176 -0.008
1 -0.097 -0.066 0.341
1 0.027 -0.135 0.325
16 0.0953 -0.3605 0.2575
8 -0.0469 -0.4174 0.2116
8 0.2185 -0.4328 0.3718
8 0.1185 -0.3211 0.0456
8 0.0951 -0.2702 0.4089 -
I think I got it but I do not know how to explain it. I set it up as below after lots of trial and error
MgNH4SO4*6H2O from Mg, Acta Crystallographica 17 (1964) 1478-1479
CRYSTAL
1 0 0
P 1 21/a 1
9.383 12.669 6.220 107.05
20
7 0.1321 0.3509 0.3611
1 0.058 0.337 0.225
1 0.208 0.305 0.394
1 0.095 0.344 0.487
1 0.174 0.421 0.346
12 0 0 0
8 0.1603 -0.1094 -0.0307
8 0.1685 0.1042 0.1656
8 -0.0017 -0.0687 0.2986
1 0.2 0.091 0.317
1 0.227 0.134 0.116
1 0.252 -0.096 0.059
1 0.143 -0.176 -0.008
1 -0.097 -0.066 0.341
1 0.027 -0.135 0.325
16 0.0953 -0.3605 0.2575
8 -0.0469 -0.4174 0.2116
8 0.2185 -0.4328 0.3718
8 0.1185 -0.3211 0.0456
8 0.0951 -0.2702 0.4089
EOS
RANGE
0.94 1.06 8
PREOPTGEOM
MAXCYCLE
500
END
BASISSET
pob-TZVP-rev2
DFT
B3LYP-D3
XLGRID
END
TOLINTEG
8 8 8 8 16
SHRINK
4 4
BIPOSIZE
41202400
EXCHSIZE
41202400
END -
Hi,
I think it was just a syntax problem on the way the symbol of the space group was inputted. This is how it should look like in this case:
TEST CRYSTAL 1 0 0 P 21/A 9.383 12.669 6.220 107.05 20 7 0.1321 0.3509 0.3611 1 0.058 0.337 0.225 1 0.208 0.305 0.394 1 0.095 0.344 0.487 1 0.174 0.421 0.346 12 0 0 0 8 0.1603 -0.1094 -0.0307 8 0.1685 0.1042 0.1656 8 -0.0017 -0.0687 0.2986 1 0.2 0.091 0.317 1 0.227 0.134 0.116 1 0.252 -0.096 0.059 1 0.143 -0.176 -0.008 1 -0.097 -0.066 0.341 1 0.027 -0.135 0.325 16 0.0953 -0.3605 0.2575 8 -0.0469 -0.4174 0.2116 8 0.2185 -0.4328 0.3718 8 0.1185 -0.3211 0.0456 8 0.0951 -0.2702 0.4089 CIFPRT TESTGEOM END ENDFor future reference, the syntax for space group symbols in CRYSTAL is discussed at pages 22-23 of the CRYSTAL23 User's Manual (see screenshot below):

We ran a test and the structure built from CRYSTAL looks reasonable:
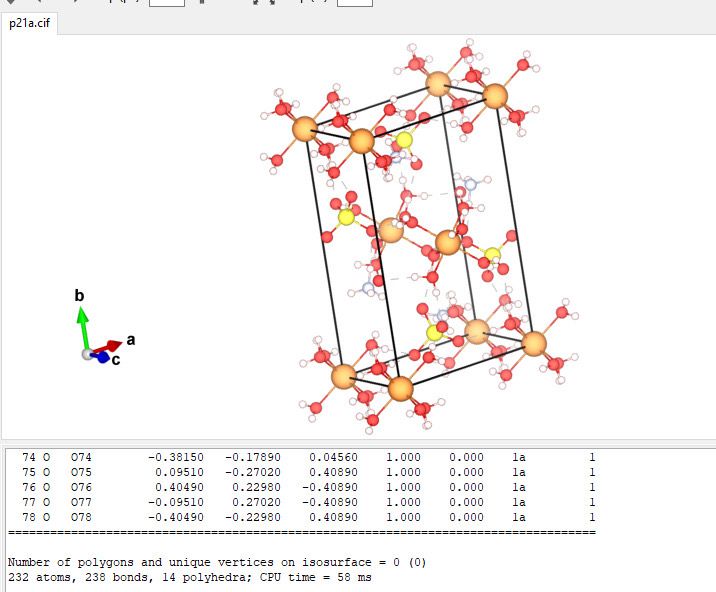
Hope this helps,
Alessandro Erba
Professor of Physical Chemistry
Department of Chemistry, University of Torino
[email protected] -
My initial confusion was that I entered
0 0 0
14and that led to problems. Today I rectified, right before your email as
CRYSTAL
1 0 0
P 1 21/a 1 -
Good! The clean way is:
CRYSTAL
1 0 0
P 21/AI guess the extra "1"s you put are safe as they correspond to the identity operator.
