extract asymmetric fragment
-
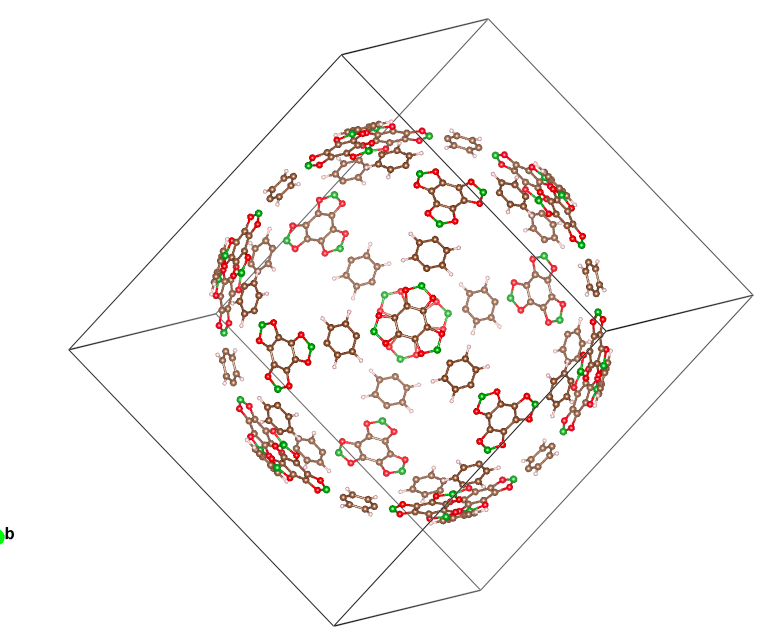
I have this 600 atom Ih symmetry molecular cage. I am seeking help extracting the asymmetric unit from it to enter into CRYSTAL so I can use Ih symmetry. If anybody could help me with it, I would appreciate it.
JB
-
Hi,
Jefferson Maul has managed to generate this .cif file with 4 symmetry operators. As you suggest, there are probably more but this is what he could extract so far.
Beautiful system by the way: looks like a Christmas tree bauble!

Alessandro Erba
Professor of Physical Chemistry
Department of Chemistry, University of Torino
[email protected] -
very grateful
-
Dear Jonas,
I tried using pymatgen to extract the point-symmetry information from your .xyz file (see the Python script below):
from pymatgen.core import Molecule from pymatgen.symmetry import analyzer bigstructure = Molecule.from_file("yourfile.xyz") PGstructure = analyzer.PointGroupAnalyzer(bigstructure) sym_mol = PGstructure.get_equivalent_atoms() print(sym_mol["eq_sets"])This returns a Python data structure containing the symmetry-irreducible sets of atoms (only 6 for this system, which is indeed of Ih point group!).
When preparing the CRYSTAL input, be careful with the orientation of your asymmetric unit. In my case, for example, I had to change the sign of the x and y coordinates to make the symmetry consistent with CRYSTAL’s conventions.
Icosahedral point groups are available in CRYSTAL (Ih is point group number 47 in CRYSTAL), so the input fort this molecular cage reads:Symm. structure MOLECULE 47 6 8 2.605032231 -11.914271806 11.762689798 6 4.344538598 15.664236912 4.366798884 6 3.427479683 14.428996321 8.326818862 6 -8.906580884 1.370529673 14.411150640 1 2.632960331 14.962480562 7.832453804 5 -8.776255231 -2.916256114 14.200279302 COORPRT TESTGEOM ENDaerba Christmas is already in the air indeed!
Davide Mitoli, PhD
Postdoctoral Researcher
Department of Chemistry - University of Torino
V. Giuria 5, 10125 Torino (Italy)
https://github.com/davidemitoli


