Alessandro, the setback is temporary. What I really appreciate is that you gave CRYSTAL more life via user support than it ever had. I am very grateful for it
Jonas Baltrusaitis
@job314
Posts
-
pov-TZVP vs old school basis sets -
Error in RESTART of FREQCALC calculationHI all, admittedly I am running these large frequency jobs and they run out of queue and I can't restart them, there is always some problem, this one is io error, hard to troubleshoot
-
corrupted size vs. prev_size while consolidatingAnother one. I emailed separately CRYSTAL support some time ago about these, Alessandro is aware of these
INPUT.d12
EOSerror.out -
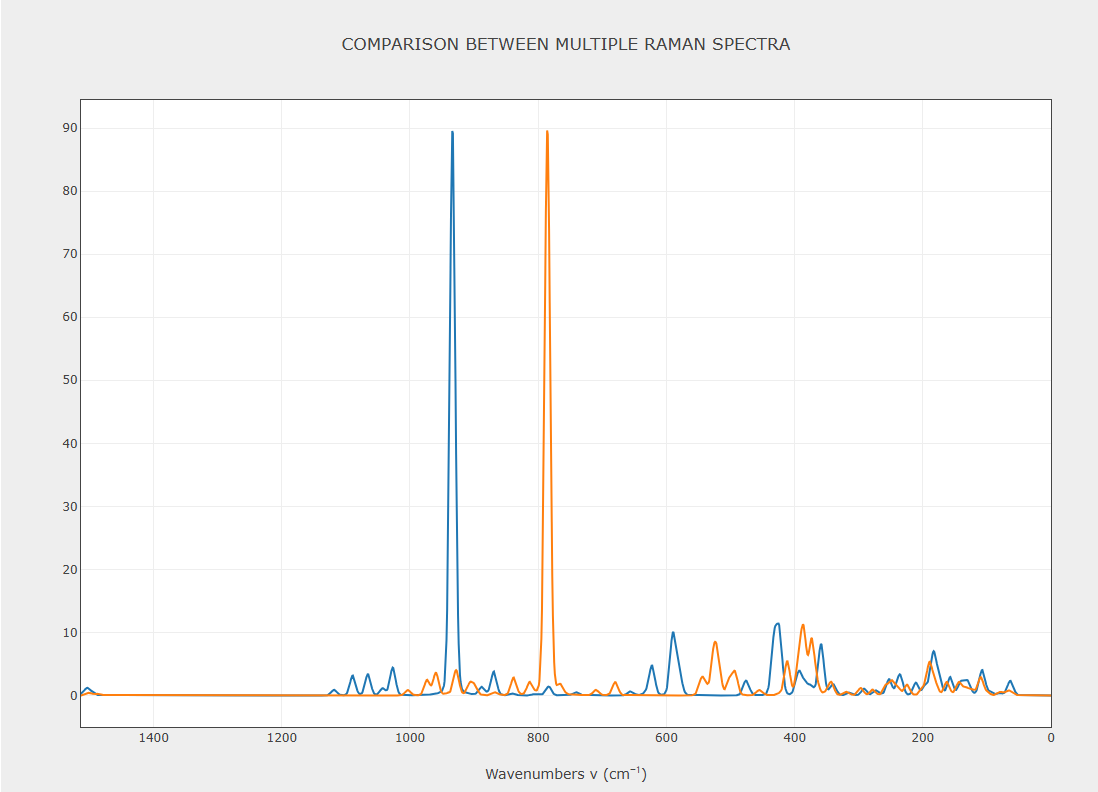
pov-TZVP vs old school basis setsMy current activity on this forum is due to the need to calculate Raman spectra. This one is not a problem but more of an observation. For the longest time I thought that CRYSTAL had a basis set problem where basis sets were seemingly randomly optimized and there were many falvors of them and they wer nore standartized in terms of contractions (e.g. 6-31G*). I was very happy to find out that standard basis sets by Peintinger were created, e.g. pob-TZVP which I can simply specify with BASISSET. So in all of my recent work on Raman spectra I used them for simplicity and uniformity.
Fast forward, my calculated Raman peaks just do not have the right position. I understand there will be an intrinsic error in the calculated peak positions, but the peaks ere 150-200 cm-1 too low for a sulfate ion. So I looked back at CRYSTAL references that did Raman including Roberto's and noticed at that time they still used these old school basis sets. I was curious and for B3LYP-D3 (I have molecular crystals or ionic crystals) with pob-TZVP I recalculated everything with these large 8611 basis sets. Much to my dismay, these old school basis sets yielded calculated Raman peaks in the vicinity of the experimental, e.g. 150-200 cm-1 higher than pob-TZVP with everything else in the input the same.
This is not a cimplaint, I understand that frequency outcome will depend on the calculation setup, e.g. functional and basis set, but why would pob-TZVP be so spectacularly bad at these Raman vibrations?
See attached, blue is 86111 old school basis set and orange is pob-TZVP basis set - I have much more data with pob-TZVP (unfortunately).
Experimental SO4 vibration is around 920 cm-1 in this particular mineral