Also, is there a way to restart calculations since I want to run for a series of increasing temperatures?
Jonas Baltrusaitis
@job314
Posts
-
Stability calculations -
Stability calculationsSorry, Alessandro, been reading over the weekend the paper and the manual. How do I perform dynamic stability in terms of vibrational frequencies as a function of temperature? There seem to be various procedures involved on page 265 of the manual but it is not apparent to me how to proceed with the optimized molecular fullerene. Can you please provide input example for several temperatures?
-
Stability calculations -
Stability calculationsAlessandro, I read your JPhysChem Lett paper from 2020. It discusses the thermo-elastic constant calculation. Can that be used as an argument, e.g., calculations at various temps would result in some decreasing elastic constant value until it approaches zero?
Additionally, any of these tensor calculations applicable to 0D systems? I have molecular cages, such as fullerene, for example -
Stability calculationsExellent. Can that be done at finite temperature? in the example I attached, they did MD simulations at increasing temps and at some point the deviation took place suggesting say at 1000 K system undergoes mechanical stability change, perhaps change in phase. Can Elastic tensors be used for that, e.g. can elastic tensors be calculated at various temperatures and see at which point they become negative to judge something about he instability temperature? Cause that woud work for me wel. Can elastic tensors be calculated at various temperatures or that concept does not exist?
-
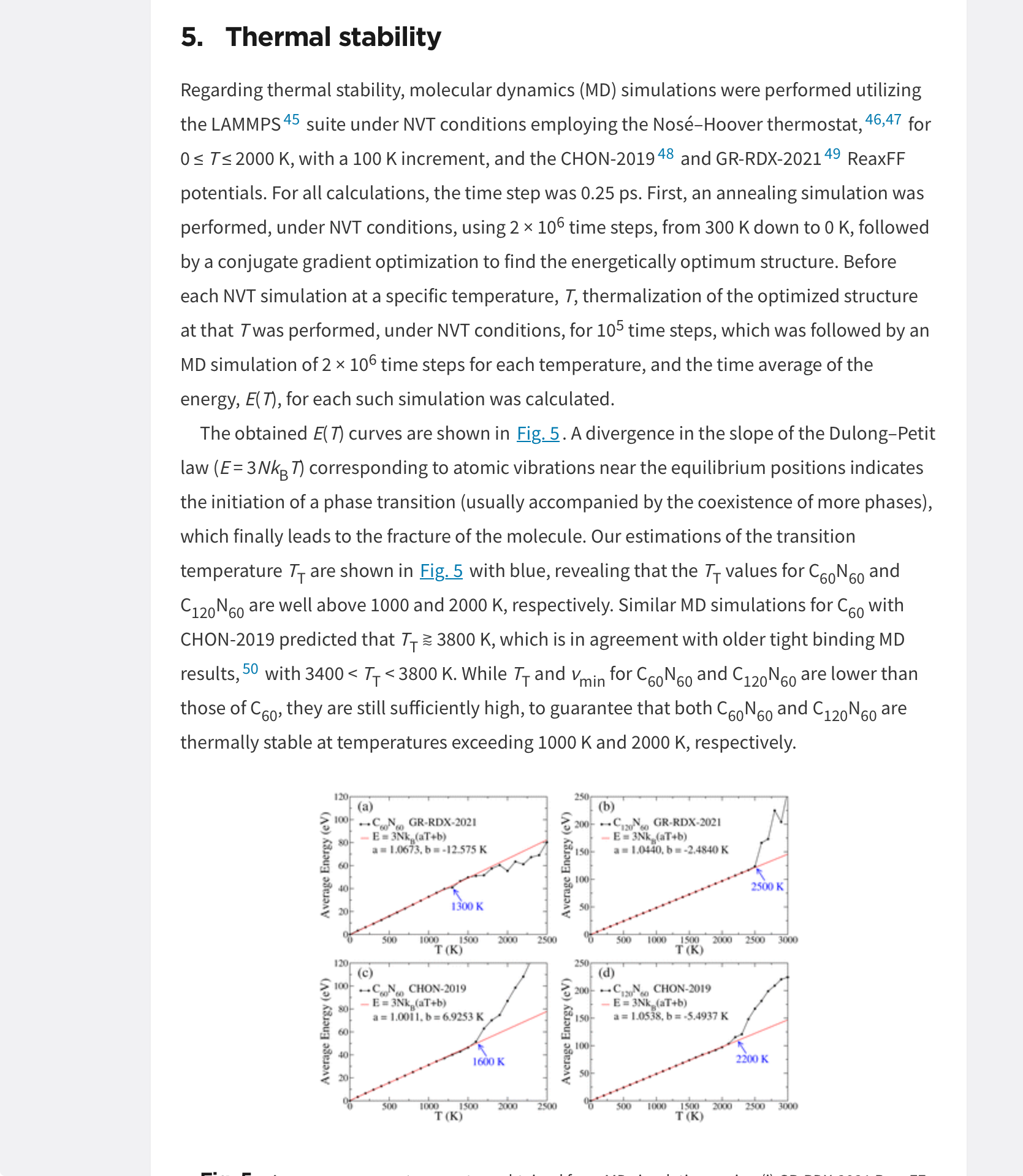
Stability calculationsI have a paper revisions requested and they asked for stability calculations. I found a similar paper, they used MD calculations. Thier plot are shown below and they talk about atomic vibrations and Dulong-Petit law. I was wondering if that has anything to do with lattice dynamics of any sort I can calculate in CRYSTAL to measure this equilibrium point before phase transition occurs? So not necessarily the method in the paper below, but something similar that show molecular stability with a temperature in a similar fashion?
Thank you
-
phonon dispersion in thermo -
phonon dispersion in thermo -
one zero frequency mode is missing -
one zero frequency mode is missingI am used to seeing 3 zero frequency modes in the crystal frequency calculation output. This urea crystal calculation has only two - I was wondering why and can I use this outcome for thermodynamics calculations with this missing mode
MODES EIGV FREQUENCIES IRREP IR INTENS RAMAN (HARTREE**2) (CM**-1) (THZ) (KM/MOL) 1- 2 -0.1181E-19 0.0000 0.0000 (E ) A ( 0.00) A 3- 3 0.2094E-19 0.0000 0.0000 (B2 ) A ( 0.00) A 4- 4 0.1254E-06 77.7189 2.3300 (B1 ) I ( 0.00) A 5- 5 0.2460E-06 108.8462 3.2631 (A2 ) I ( 0.00) I 6- 7 0.3349E-06 127.0111 3.8077 (E ) A ( 0.00) A 8- 8 0.4355E-06 144.8406 4.3422 (A1 ) I ( 0.00) A 9- 10 0.6407E-06 175.6769 5.2667 (E ) A ( 0.00) A 11- 12 0.1108E-05 230.9837 6.9247 (E ) A ( 0.00) A 13- 13 0.4844E-05 483.0526 14.4816 (B1 ) I ( 0.00) A 14- 14 0.6693E-05 567.8172 17.0227 (A1 ) I ( 0.00) A 15- 16 0.6926E-05 577.5886 17.3157 (E ) A ( 0.00) A 17- 17 0.7310E-05 593.3990 17.7897 (B2 ) A ( 0.00) A 18- 19 0.7970E-05 619.6054 18.5753 (E ) A ( 0.00) A 20- 20 0.8638E-05 645.0548 19.3383 (A2 ) I ( 0.00) I 21- 21 0.1067E-04 717.0169 21.4956 (B1 ) I ( 0.00) A 22- 22 0.1286E-04 787.0371 23.5948 (A2 ) I ( 0.00) I 23- 24 0.1322E-04 798.0946 23.9263 (E ) A ( 0.00) A 25- 26 0.1420E-04 826.9504 24.7914 (E ) A ( 0.00) A 27- 27 0.2232E-04 1036.9495 31.0870 (B2 ) A ( 0.00) A 28- 28 0.2251E-04 1041.3084 31.2176 (A1 ) I ( 0.00) A 29- 30 0.2535E-04 1105.1276 33.1309 (E ) A ( 0.00) A 31- 31 0.2780E-04 1157.1639 34.6909 (B2 ) A ( 0.00) A 32- 32 0.3003E-04 1202.7304 36.0569 (A1 ) I ( 0.00) A 33- 34 0.4803E-04 1521.0625 45.6003 (E ) A ( 0.00) A 35- 35 0.4892E-04 1535.0190 46.0187 (A1 ) I ( 0.00) A 36- 36 0.5334E-04 1602.8976 48.0537 (B2 ) A ( 0.00) A 37- 38 0.5717E-04 1659.4784 49.7499 (E ) A ( 0.00) A 39- 39 0.5840E-04 1677.2645 50.2831 (A1 ) I ( 0.00) A 40- 40 0.6098E-04 1713.8726 51.3806 (B2 ) A ( 0.00) A 41- 42 0.2380E-03 3386.0936 101.5125 (E ) A ( 0.00) A 43- 43 0.2424E-03 3416.7391 102.4313 (A1 ) I ( 0.00) A 44- 44 0.2438E-03 3426.7219 102.7305 (B2 ) A ( 0.00) A 45- 46 0.2612E-03 3547.0252 106.3371 (E ) A ( 0.00) A 47- 47 0.2614E-03 3548.4246 106.3791 (A1 ) I ( 0.00) A 48- 48 0.2657E-03 3577.3918 107.2475 (B2 ) A ( 0.00) A -
phonon dispersion in thermoDear all, what is the purpose of the phonon dispersion calculations according to the tutorial? What is the effect we are seeing, going beyond the gamma point?
https://tutorials.crystalsolutions.eu/tutorial.html?td=thermo&tf=thermo2#phonon
More importantly, how do I set it up properly? What size SCELPHONO should I be after? Is the point to have all of my crystals about the same atoms in the generated SCELPHONO for appropriate comparison? I was perusing the tutorial but I could not quite understand the purpose or the correct setup of SCELPHONO
thank you
Jonas
-
D infinity h symmetry -
D infinity h symmetry -
extract asymmetric fragment -
SCANMODE io error Read_int_1dSo it turns out having fort.13 and fort.20 is not optional for restart. Now I still need help. I optimizing this 600 atom cluster with Ih and can see that it is not minimum, there are negatives - this is why I am scanning. The scan does not return a reasonable local minimum. I only scanned negative branch but I assume they are symmetrical.
I would appreciate any helphttps://www.dropbox.com/t/5jgMXmCEPNLGHpAY
PS. I would also like to ask developers to allow uploading compressed files to this forum
SSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS SCAN ALONG NORMAL MODES STARTING POINT: -20 ENDING POINT: 20 STEP: 0.20000 (THE STEP IS GIVEN AS TIMES OF THE CLASSICAL AMPLITUDE AT THE QUANTUM GROUND STATE ENERGY. THE MAX ATOMIC DISPLACEMENT IN THE STEP IS GIVEN IN BOHR WITHIN SQUARE BRACKETS) MODE(CM-1) DISPLAC TOTAL ENE(DFT)(AU) CLASSICAL HARM ENE(AU) NCYC DE [MAX DISP -NATOM-] 1( -11.5) [ 0.016 - 512-] -4.0000 -0.2201822589948E+05 -0.2201822692117E+05 15 -0.7E-09 -3.8000 -0.2201822596504E+05 -0.2201822688013E+05 15 -0.5E-09 -3.6000 -0.2201822602580E+05 -0.2201822684119E+05 15 -0.5E-09 -3.4000 -0.2201822608201E+05 -0.2201822680436E+05 16 0.3E-10 -3.2000 -0.2201822613398E+05 -0.2201822676964E+05 14 0.1E-08 -3.0000 -0.2201822618174E+05 -0.2201822673702E+05 17 0.1E-10 -2.8000 -0.2201822622563E+05 -0.2201822670650E+05 17 -0.7E-10 -2.6000 -0.2201822626589E+05 -0.2201822667809E+05 18 0.2E-10 -2.4000 -0.2201822630238E+05 -0.2201822665178E+05 16 -0.9E-10 -2.2000 -0.2201822633530E+05 -0.2201822662758E+05 17 0.0E+00 -2.0000 -0.2201822636488E+05 -0.2201822660548E+05 16 0.4E-10 -1.8000 -0.2201822639128E+05 -0.2201822658549E+05 16 -0.4E-10 -1.6000 -0.2201822641463E+05 -0.2201822656760E+05 15 -0.4E-10 -1.4000 -0.2201822643453E+05 -0.2201822655181E+05 17 0.4E-10 -1.2000 -0.2201822645245E+05 -0.2201822653813E+05 14 0.1E-10 -1.0000 -0.2201822646714E+05 -0.2201822652656E+05 15 -0.5E-10 -0.8000 -0.2201822647907E+05 -0.2201822651709E+05 14 -0.4E-10 -0.6000 -0.2201822648831E+05 -0.2201822650972E+05 15 -0.4E-10 -0.4000 -0.2201822649498E+05 -0.2201822650446E+05 14 0.3E-10 -0.2000 -0.2201822649893E+05 -0.2201822650130E+05 12 -0.6E-10 0.0000 -0.2201822650025E+05 -0.2201822650025E+05 CENTRAL POINT -
SCANMODE io error Read_int_1d -
SCANMODE io error Read_int_1d -
SCANMODE io error Read_int_1dDear all, I am getting the IO error when scanning modes and no idea why. I copied HESSFREQ.DAT and FREQINFO.DAT files into the scan directory. I would appreciate your help
Unfortunately, it is a large system and I can't upload the files here, I will move them via Dropbox, see the link below
https://www.dropbox.com/t/uVxYAZdbhCFpM79K
SSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS SCAN ALONG NORMAL MODES STARTING POINT: -10 ENDING POINT: 10 STEP: 0.40000 (THE STEP IS GIVEN AS TIMES OF THE CLASSICAL AMPLITUDE AT THE QUANTUM GROUND STATE ENERGY. THE MAX ATOMIC DISPLACEMENT IN THE STEP IS GIVEN IN BOHR WITHIN SQUARE BRACKETS) MODE(CM-1) DISPLAC TOTAL ENE(DFT)(AU) CLASSICAL HARM ENE(AU) NCYC DE [MAX DISP -NATOM-] 1( -8.5) [ 0.030 - 390-] io error Read_int_1d 300 -1 Abort(1) on node 0 (rank 0 in comm 0): application called MPI_Abort(MPI_COMM_WORLD, 1) - process 0 -
extract asymmetric fragmentI have this 600 atom Ih symmetry molecular cage. I am seeking help extracting the asymmetric unit from it to enter into CRYSTAL so I can use Ih symmetry. If anybody could help me with it, I would appreciate it.
JB
-
SCANMODE problemHi Aleks, I tried but ended up just using LDREMO keyword
Another superstrange thing (that of course was not happening yesterday), that I can't seem to specify initial and final step anymore properly, it divides it by 10... Bizzare. I now have to multiply initial step by 10 (-1.6 start I want I need to specify as 16!) so it startes at -1.6...My input is:
EXTERNAL FREQCALC RESTART SCANMODE 1 -16 0 0.1 1 END ENDSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSSS SCAN ALONG NORMAL MODES STARTING POINT: -16 ENDING POINT: 0 STEP: 0.10000 (THE STEP IS GIVEN AS TIMES OF THE CLASSICAL AMPLITUDE AT THE QUANTUM GROUND STATE ENERGY. THE MAX ATOMIC DISPLACEMENT IN THE STEP IS GIVEN IN BOHR WITHIN SQUARE BRACKETS) MODE(CM-1) DISPLAC TOTAL ENE(DFT)(AU) CLASSICAL HARM ENE(AU) NCYC DE [MAX DISP -NATOM-] 1( -62.7) [ 0.028 - 3-] -1.6000 -0.1610014638959E+04 -0.1610015023766E+04 18 -0.1E-07 -1.5000 -0.1610014685887E+04 -0.1610014979469E+04 29 0.5E-06 -1.4000 -0.1610014719434E+04 -0.1610014938029E+04 13 -0.9E-07 -1.3000 -0.1610014740251E+04 -0.1610014899447E+04 13 -0.2E-06 -1.2000 -0.1610014750671E+04 -0.1610014863723E+04 21 -0.9E-08 -1.1000 -0.1610014754239E+04 -0.1610014830856E+04 15 -0.4E-06 -1.0000 -0.1610014749993E+04 -0.1610014800848E+04 28 0.1E-07 -0.9000 -0.1610014740870E+04 -0.1610014773698E+04 23 0.1E-05