Error in RESTART of FREQCALC calculation
-
HI all, admittedly I am running these large frequency jobs and they run out of queue and I can't restart them, there is always some problem, this one is io error, hard to troubleshoot
-
Hi,
I am quite familiar with that error message myself: it pops up when the fort.9 unit provided upon the restart is the wrong one (i.e. does not match with the current calculation).
Let me make a general comment on how to best approach these frequency+intensity calculations with CRYSTAL based on my experience.
I noticed that you tend to use the PREOPTGEOM option within FREQCALC, and to compute infrared and Raman intensities, all in one shot. Now, this is a very nice feature of CRYSTAL that allows you to run a single job where everything is fully automated: the structure gets optimized, the numerical Hessian computed and diagonalized, and Born and Raman tensors computed. Not many programs can do that, to the best of my knowledge. At the same time, while this is very convenient if you can indeed run everything in a single job, it complicates things if you then need to do a restart from a previous incomplete calculation.
If you envisage that this could be the case (maybe because of a wall clock limit on the cluster) then it is preferable (to put it mildly) to do things step by step:
- You first run a geometry optimization with something like:
[initial geometry] OPTGEOM END- You prepare a new input file where you insert the optimized geometry from the previous run as a starting point, and perform a frequency calculation, with something like:
[optimized geometry] FREQCALC END- If this calculation stops, you can now restart it easily with:
[optimized geometry] FREQCALC RESTART ENDby providing the required restart files (FREQINFO.DAT and fort.13 from the first frequency calculation, and fort.9 from the geometry optimization, to be renamed fort.20 in the new scratch folder).
- Once the restart is done and the frequency calculation is complete, you can compute the intensities, with something like:
[optimized geometry] FREQCALC RESTART INTENS INTRAMAN INTCPHF ENDCPHF ENDPersonally, I always do things separately, running first the geometry optimization, then the Hessian (i.e. harmonic frequency) calculation, then the intensities through CPHF/KS in three separate jobs.
Hope this clarifies things a little,
-
It is tough, Alessandro. Restarting frequency calculations (or just running them) is unpredictable. It runs normally, then if it stops out of time and is restarted, many times it does not run for one reason or the other - see a random restart abort below. It is tough, perhaps I will learn something by doing them but I now normally just start form scratch
DIIS TEST: 0.85073E-04 AT CPHF CYCLE 9 - DIIS ACTIVE - HISTORY: 9 CYCLES
CYCLE 9 ALPHA 278.474328 EPSILON 2.093136 DELTA 4.8487E-04
forrtl: severe (256): unformatted I/O to unit open for formatted transfers, unit 85, file /dev/null
Image PC Routine Line Source
Pcrystal 0000000007374206 Unknown Unknown Unknown
Pcrystal 0000000001BA179E Unknown Unknown Unknown
Pcrystal 0000000000A8038B Unknown Unknown Unknown -
Alessandro, to prove the point how difficult these restarts are, I copied FREQINFO.DAT from the first frequency calculation, fort.13 unit, and fort.9, renamed fort.20 in the new scratch folder, grabbed the last optc file from the converged geometry optimization. Tried the restart with GUESSP since I am getting conducting state otherwise. Right away I have problems where my old and new vectors via GUESSP are different. why?
INFORMATION FROM INTEGRAL EVALUATION
RESTART FROM A PREVIOUS RUN FOCK MATRIX - DEP ACTIVE
NUMBER OF COUPLE SETS (NEW, OLD, FOUND): 50745 15118 15039
NUMBER OF IRREDUCIBLE G VECTORS : 71990 64543 63141 -
Alessando, can I send you the files above so you can please try a restart? My restarts end up being conducting state and do not converge
-
Yes, please, send me all the files.
-
-
Whew... At least you got it... It is uncanny how a normally job, aborted, can't be restarted since it can't converge SCF all of a sudden...
-
Hi,
I have run some calculations on your case following my step-by step recipe (as described at https://forum.crystalsolutions.eu/post/339, which I have now edited to include the intensity step), and the restart now works just fine (i.e. without the annoying "possibly conducting state", and with a nice convergence of the further SCFs upon restart).
This is what I did:
-
I took the optimized geometry from your original output file and I created a new input file for a single-point calculation. I ran it and obtained the wavefunction external file (i.e. fort.9 unit);
-
I prepared an input file to run the harmonic frequency calculation, I ran it and I killed it in the middle of the construction of the Hessian. From this incomplete frequency calculation, I obtained the FREQINFO.DAT file and the external unit with the density matrix, i.e. fort.13 unit;
-
I prepared an input file to restart the frequency calculation and provided the FREQINFO.DAT and fort.13 files from the previous step and the fort.9 (renamed as fort.20) from the first step, and ran it. The calculation of the Hessian restarted correctly, with the new SCFs converging nicely (see the attached SCFOUT.LOG file). I then stopped the calculation not to use too much compute power on my cluster.
If you follow this step-by-step process you'll be able to safely restart your frequency calculations.
Hope this helps,
Alessandro Erba
Professor of Physical Chemistry
Department of Chemistry, University of Torino
[email protected] -
-
Hey, just to add onto this, I'm getting a similar error where the SCF won't wouldn't converge upon restart - but only occurs with certain structures.
When I do with the restart with CRYSTAL17, it works perfectly - it's odd that it only happens on restart because when I lower the criteria, it doesn't need a restart and completes the calculation - but I can't keep lowering the tolerance criteria when I encounter this issue.CRYSTAL17 succesful restart:
BLACTO_PBE_D3_AhTZVP_FREQ_4_4-53910034 - CRYSTAL17 success..out
CRYSTAL23 unsucessful restart + SCFOUT:
BLACTO_PBE_D3_AhTZVP_FREQ_4_4-53437836.out
SCFOUT.LOGAny ideas around this?
I've also included a plot of the DETOT from the SCFOUT.LOG files - where the successful one was from CRYSTAL17
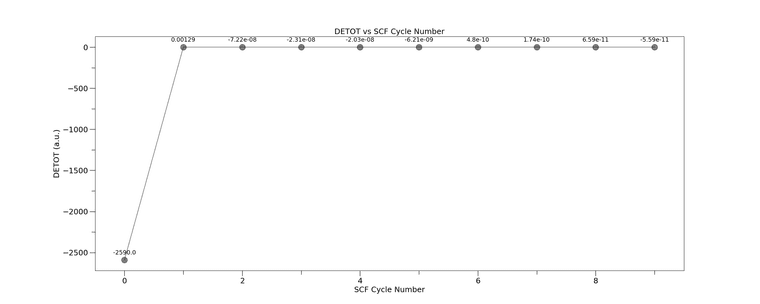
The then rest from CRYSTAL23
detot_vs_cycle_CRYSTAL23_FAIL_CYC0tomax.png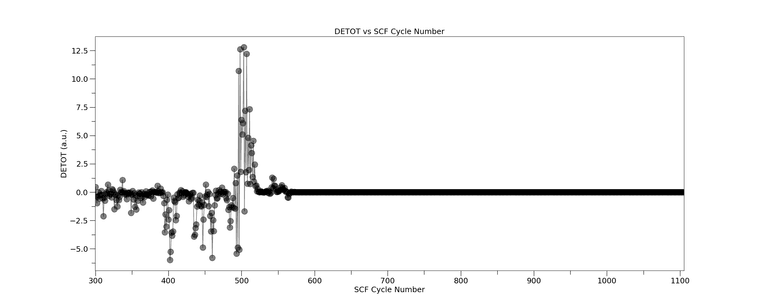
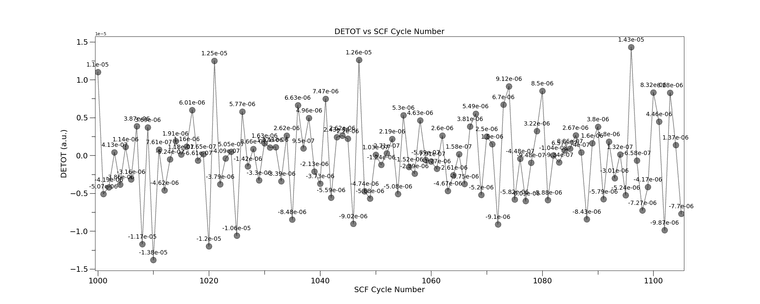
Our HPC platform provider said it was not an issue on their end and advised to seek support from the CRYSTAL team. Just to add on, our HPC platform provider also said I can't use the CRYSTAL17 anymore because they can't build the architecture on their new upgrade which, honestly, I don't really understand why.
Happy to discuss,
Peter -
Hi,
By the looks of it, it may be due to some mismatch in the restart units. I am happy to run some tests. Could you share your input files for this case?
-
Sure, I've uploaded the files input files in this link because it exceeded the file size limit to upload them in this comment:
https://otagouni-my.sharepoint.com/:f:/g/personal/rempe782_student_otago_ac_nz/EmtG3VsEJYJArghuK_IF1x0BvJNBiYsE1mrbQELFWOE1-A?e=NhXhQsUsed the same inputs (.d12, fort.9, fort.13, fort.20, FREQINFO.DAT) for to do the frequency calculation restarts for CRYSTAL17 and 23
Thanks heaps
-
aerba said in Error in RESTART of FREQCALC calculation:
Hi,
I have run some calculations on your case following my step-by step recipe (as described at https://forum.crystalsolutions.eu/post/339, which I have now edited to include the intensity step), and the restart now works just fine (i.e. without the annoying "possibly conducting state", and with a nice convergence of the further SCFs upon restart).
This is what I did:
-
I took the optimized geometry from your original output file and I created a new input file for a single-point calculation. I ran it and obtained the wavefunction external file (i.e. fort.9 unit);
-
I prepared an input file to run the harmonic frequency calculation, I ran it and I killed it in the middle of the construction of the Hessian. From this incomplete frequency calculation, I obtained the FREQINFO.DAT file and the external unit with the density matrix, i.e. fort.13 unit;
-
I prepared an input file to restart the frequency calculation and provided the FREQINFO.DAT and fort.13 files from the previous step and the fort.9 (renamed as fort.20) from the first step, and ran it. The calculation of the Hessian restarted correctly, with the new SCFs converging nicely (see the attached SCFOUT.LOG file). I then stopped the calculation not to use too much compute power on my cluster.
If you follow this step-by-step process you'll be able to safely restart your frequency calculations.
Hope this helps,
thank you and I am trying to do that
-
-
PeterRemoto I just started running some tests on this case. I will keep you posted on how it goes.
-
PeterRemoto I started drafting a reply but encountered problems in uploading the files. Will try later.
Alessandro Erba
Professor of Physical Chemistry
Department of Chemistry, University of Torino
[email protected] -
aerba I saw it earlier and glad that a nice solution was found. Thank you so much - this has actually stumped me months ago before getting busy with other projects. I was wondering though, any thoughts on why this frequency restart issue is happening to a handful of structures for CRYSTAL23 but then the restarts worked fine for 17?
-
PeterRemoto I have tried to restart your calculation with CRYSTAL23 following my step-by-step recipe (described at https://forum.crystalsolutions.eu/post/339) and it worked.
This is what I did:
-
I prepared an input file to run the harmonic frequency calculation, without intensities, I ran it and I killed it in the middle of the construction of the Hessian. From this incomplete frequency calculation, I obtained the FREQINFO.DAT file (I can not upload it here as it exceeds the maximum allowed file size), the external unit with the density matrix, i.e. fort.13 unit, and the external unit with the wavefunction, i.e. fort.9;
-
I prepared an input file to restart the frequency calculation and provided the FREQINFO.DAT, fort.13 and the fort.9 (renamed as fort.20) files from the previous step, and ran it. The calculation of the Hessian restarted correctly, with the new SCFs converging nicely (see the attached SCFOUT.LOG file). I then stopped the calculation not to use too much compute power on my cluster.
If you follow this step-by-step process you'll be able to safely restart your frequency calculations with CRYSTAL23.
Hope this helps,
-
-
PeterRemoto At the moment I am afraid I do not have an explanation for the difference you experienced between CRYSTAL17 and CRYSTAL23 as I am unable to reproduce the erratic behavior with CRYSTAL23.
Alessandro Erba
Professor of Physical Chemistry
Department of Chemistry, University of Torino
[email protected] -
aerba I've noticed for my systems, these SCF convergence issues appear when I try to compute for intensities in a restart job. Then when removing intensities, they converge fine and progress forward.
Is it possible to finish the frequency calculations first and then compute for the Raman intensities afterwards? Would that be a work around? Maybe with RAMANREA - because I have the TENS_RAMAN.DAT, but it just gets stuck at some point during HHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHH
FORCE CONSTANT MATRIX - NUMERICAL ESTIMATE
HHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHHH
and doesn't progress with restarts
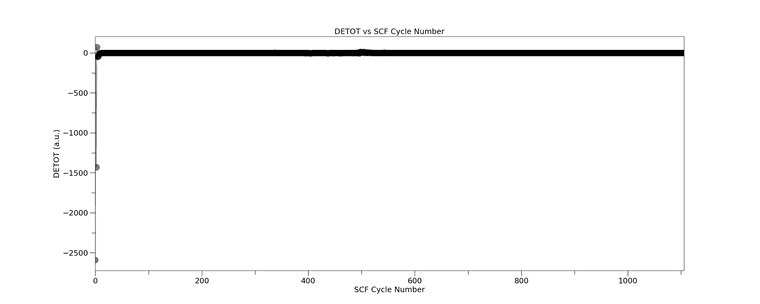{kind=link}