Is it necessary to add the keyword NOSYMADA when using a user-defined anisotropic k-point grid?
-
Hello, I have a question regarding the use of the NOSYMADA keyword in CRYSTAL.
While reading the CRYSTAL manual, I came across the note that “User defined anisotropic net is not compatible with SABF (Symmetry Adapted Bloch Functions). See NOSYMADA, page 122.” (page 126).
However, I noticed that in the following example: https://wiki.aalto.fi/pages/viewpage.action?pageId=180298659, the author performed geometry optimization for Sr₅(BO₃)₃H using an anisotropic k-point grid, but did not include the NOSYMADA keyword.
I also did a test myself: I defined an anisotropic k-point mesh (1×5×5) and did not use NOSYMADA. In the .out file, I still see the line:
“NUMBER OF SYMMETRY OPERATORS : 2”,
which suggests that symmetry was not automatically disabled despite using an anisotropic grid.So my question is:
When using a user-defined anisotropic k-point grid in CRYSTAL, is it necessary to explicitly include the NOSYMADA keyword? -
Hi Yachao_su,
From my experience (at least with CRYSTAL23), using anisotropic shrinking factors does not automatically enforce the use of the NOSYMADA keyword. Thus, symmetry is still retained unless you explicitly specify it.
So, I believe the statement in the manual might be outdated.
Giacomo Ambrogio, PhD Student
Department of Chemistry - University of Torino
V. Giuria 5, 10125 Torino (Italy) -
GiacomoAmbrogio Thank you for your reply. When performing k-point convergence tests with anisotropic meshes, I noticed that the inclusion or omission of the NOSYMADA keyword — which controls whether symmetry is applied — results in slight differences in total energy.
For the same system, I obtained two different convergence outcomes:
With NOSYMADA, the convergence was reached with a 1×5×5 grid;
Without NOSYMADA, the result suggested 1×7×7.
Since the use of symmetry affects the total energy, my questions are:
Which setup yields a more reliable and physically meaningful result for k-point convergence in this case?

-
Can you please share the input file?
Giacomo Ambrogio, PhD Student
Department of Chemistry - University of Torino
V. Giuria 5, 10125 Torino (Italy) -
"Added NOSYMADA"
CZ_DBF
EXTERNAL
BASISSET
SOLDEF2MSVP
DFT
HSESOL3C
ENDDFT
NOSYMADA
SHRINK
0 5
1 5 5
TOLINTEG
7 7 7 7 14
END
Added_NOSYMADA.out"No NOSYMADA"
CZ_DBF
EXTERNAL
BASISSET
SOLDEF2MSVP
DFT
HSESOL3C
ENDDFT
SHRINK
0 5
1 5 5
TOLINTEG
7 7 7 7 14
END
No_NOSYMADA.out
CZ_DBF.cif -
This post is deleted!
-
We ran some tests regarding the inclusion of the keyword
NOSYMADAin relation to anisotropic shrinking factors, and I can confirm that we did not find any significant difference. Therefore, anisotropic shrinking is fully compatible with symmetry-adapted block functions.Taking a closer look to the data and input/output files you submitted, the convergence with respect to the shrinking factors appears to be very similar. Comparisons of calculations using the same
SHRINKvalue consistently show differences well within the default tolerance (10^-6 Ha/cell), except for one value (highlighted in red in the screenshot). In this case, the energy seems to be significantly off the trend. Could you please double-check that specific calculation?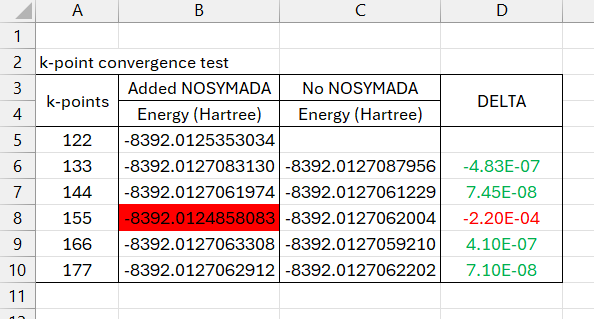
Giacomo Ambrogio, PhD Student
Department of Chemistry - University of Torino
V. Giuria 5, 10125 Torino (Italy) -
GiacomoAmbrogio Thank you for your helpful suggestion earlier! I've carefully reviewed the .out file. After adding NOSYMADA, the calculation stopped at step 9:
CYC 9 ETOT(AU) -8.392098065539E+03 DETOT -5.24E-04 tst 4.69E-06 PX 3.18E-03
== SCF ENDED - CONVERGENCE ON ENERGY E(AU) -8.3920980655389E+03 CYCLES 9
ENERGY EXPRESSION = HARTREE + FOCK EXCH * 0.00000 + (HSESOL EXCH) * 1.00000 + PBESOL CORR
As I mentioned in the input file I shared earlier, I didn’t manually modify any convergence thresholds. I was therefore surprised to see the SCF deemed converged when DETOT = -5.24E-04. From what I understand, the default SCF energy convergence criterion is 1.0E-7, so I’m wondering whether I may have missed something here. Would you mind helping me take a quick look, or point me in the right direction? Many thanks in advance!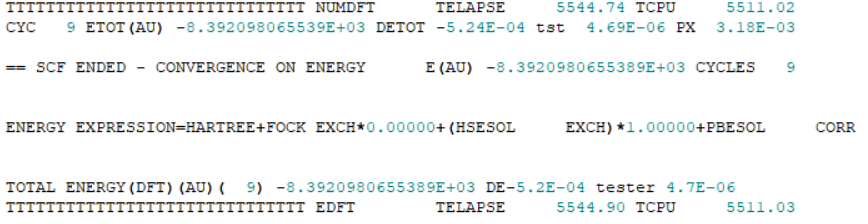 Added_NOSYMADA.out
Added_NOSYMADA.out -
Hi,
The SCF converged because at cycle 8 the energy change with respect to the previous cycle was below the default threshold of 1.0E-6:
CYC 8 ETOT(AU) -8.392097541980E+03 DETOT 8.46E-07
By inspection of this specific SCF, I would suggest to re-run it by tightening the convergence criterion a little, for instance with:
TOLDEE
8Hope this clarifies things a little
-
The default DETOT value for convergence in SCF is set to 1.0E-6 (at least in single point calculations, you can see all other settings at page 130 in the CRYSTAL user manual). This means that as soon as DETOT drops below 1.0E-6, the code considers the system to have reached convergence and stops. For technical reasons, however, the code performs one additional SCF cycle (actually, half-cycle) after this point.
This explains the behavior you're seeing. If you look at the 8th cycle, the DETOT is 8.46E-07, which triggers the SCF to stop. But clearly, the system hasn't fully converged. I'm not entirely sure why this happens, but it's likely due to parameters that are too loose... such as the thresholds for integral evaluation, DETOT, SHRINK, etc., combined with the DIIS convergence accelerator.
To tighten the SCF convergence threshold, you can use the
TOLDEEkeyword in the third input section. For example you can make the DETOT treshold 1.0E-8 like this:[...] SHRINK 0 5 1 5 5 TOLINTEG 7 7 7 7 14 TOLDEE 8 ENDAnother thing you can try is restarting the calculation using the previously obtained wavefunction as the initial guess. This will allow the code to continue the SCF after the ninth cycle and possibly move toward the correct solution. You can do this as follows:
CZ_DBF EXTERNAL BASISSET SOLDEF2MSVP DFT HSESOL3C ENDDFT GUESSP SHRINK 0 5 1 5 5 TOLINTEG 7 7 7 7 14 ENDThe important thing is to copy the wavefunction obtained from the first calculation (fort.9) into the scratch folder of the new one, renaming it to fort.20.
As a general tip, it's always a good idea to check the convergence of the calculation using:
grep DETOT output_file
This can be very helpful for spotting issues and monitoring how the code is progressing during the calculation. -
Your response is incredibly valuable—thank you so much.I suspect that the DIIS/Anderson extrapolation might have 'hit' a local minimum, so the density guess in iteration 8 happened to yield a total energy extremely close to that of the previous step, resulting in a very small ΔE.
CYC 0 ETOT(AU) -8.456120523718E+03 DETOT -8.46E+03 tst 0.00E+00 PX 1.00E+00
CYC 1 ETOT(AU) -8.390242659622E+03 DETOT 6.59E+01 tst 0.00E+00 PX 1.00E+00
CYC 2 ETOT(AU) -8.391027170997E+03 DETOT -7.85E-01 tst 3.34E-03 PX 1.13E-01
CYC 3 ETOT(AU) -8.391734802465E+03 DETOT -7.08E-01 tst 2.44E-03 PX 1.02E-01
CYC 4 ETOT(AU) -8.392064543836E+03 DETOT -3.30E-01 tst 8.38E-04 PX 4.49E-02
CYC 5 ETOT(AU) -8.392086825687E+03 DETOT -2.23E-02 tst 1.24E-04 PX 1.72E-02
CYC 6 ETOT(AU) -8.392095974656E+03 DETOT -9.15E-03 tst 2.86E-05 PX 9.66E-03
CYC 7 ETOT(AU) -8.392097542826E+03 DETOT -1.57E-03 tst 3.85E-06 PX 4.81E-03
CYC 8 ETOT(AU) -8.392097541980E+03 DETOT 8.46E-07 tst 5.41E-06 PX 3.18E-03
CYC 9 ETOT(AU) -8.392098065539E+03 DETOT -5.24E-04 tst 4.69E-06 PX 3.18E-03
While this DETOT value satisfies the default energy convergence criterion, the corresponding values suggest that the electron density had not yet fully stabilized. This aligns well with your suggestion that tightening the convergence threshold would lead to a more reliable result.
Thank you again for your guidance.

