Questions on HSE06 Band‐Gap Accuracy in CRYSTAL
-
Hi
I’m using HSE06/TZVP LOT to compute band gaps for a series of ZIF frameworks, and I’m noticing a systematic overestimation of ~0.6–1.2 eV when compared with published reference values.What level of absolute accuracy should I generally expect from CRYSTAL’s HSE06 band gaps? Are there specific numerical settings you recommend to avoid this discrepancy?
I am trying to understand what I may be doing wrong.Thanks
-
Hi,
Can you tell me if the "published reference values" you are comparing with are computational from other HSE06 implementations (if yes, which one) or experimental or computational from different functionals?
Thanks
-
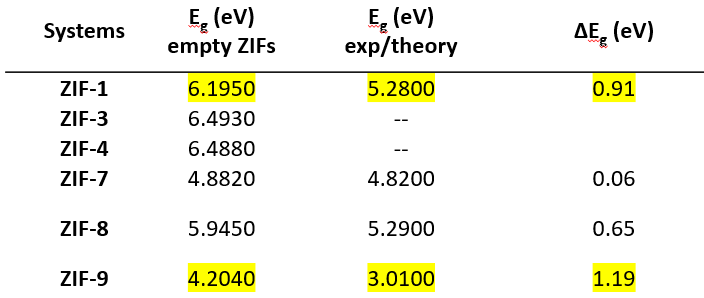
Hi,
I am following this paper which is a computational work with the HSE06 functional performed with VASP. Below, I have also attached a picture of my results for a better understanding.
Thank you for your help!
-
Hi,
Hmmm...even though the SI file of the paper you have shared does point to using HSE06, it remains not entirely clear whether its parameters were kept default or altered (for all systems together or individually for instance).
There are also differences between what different codes consider as "default" settings. For example, within the code used in the paper (VASP, nice code, no doubt), the defaults for HSE06 read (taken from https://www.vasp.at/wiki/index.php/List_of_hybrid_functionals) :
$$ \omega= 0.2\ \mathring{A} , \quad c = 0.25, \quad \text{correlation}=\text{PBE}, $$ with the first number reading the range separation parameter (omega) and the second the fraction of exact exchange used (c).Within CRYSTAL (also nice code, no doubt), these read (taken from the manual, page 138):
$$ \omega= 0.11\ a_0^{-1}, \quad c = 0.25, \quad \text{correlation}=\text{PBE}, $$ adopting the same labels.Not sure about the exact definition of units (perhaps a developer can comment if this is indeed Bohr radius as assumed?), but you can already see the subtle differences having to be taken into account when comparing between codes.
A few other thoughts worth considering:
-
In the paper, the structure was optimized with PBEsol and on top of that geometry HSE06 was applied as a single-point calculation. Not sure about the exact composition of those ZIFs, but the structural differences could play a significant role as well (planewave codes are very costly when optimizing a structure with hybrid functionals). Here is also a good read on this topic: doi.org/10.1088/2516-1075/aafc4b
-
One final small comment. Within the PAW formalism implemented in VASP, scalar relativistic effects are included in the pseudopotentials by default. No problem, cool feature, but should be taken into account when comparing results, especially for heavier elements (longer discussion found here https://blog.vasp.at/forum/viewtopic.php?t=902)
Hope this helps!
Cheers,
AleksAleksandar Živković, Scientific Assistant
Department of Earth and Environmental Sciences, LMU Munich, Germany
Crystal enthusiast -