pov-TZVP vs old school basis sets
-
My current activity on this forum is due to the need to calculate Raman spectra. This one is not a problem but more of an observation. For the longest time I thought that CRYSTAL had a basis set problem where basis sets were seemingly randomly optimized and there were many falvors of them and they wer nore standartized in terms of contractions (e.g. 6-31G*). I was very happy to find out that standard basis sets by Peintinger were created, e.g. pob-TZVP which I can simply specify with BASISSET. So in all of my recent work on Raman spectra I used them for simplicity and uniformity.
Fast forward, my calculated Raman peaks just do not have the right position. I understand there will be an intrinsic error in the calculated peak positions, but the peaks ere 150-200 cm-1 too low for a sulfate ion. So I looked back at CRYSTAL references that did Raman including Roberto's and noticed at that time they still used these old school basis sets. I was curious and for B3LYP-D3 (I have molecular crystals or ionic crystals) with pob-TZVP I recalculated everything with these large 8611 basis sets. Much to my dismay, these old school basis sets yielded calculated Raman peaks in the vicinity of the experimental, e.g. 150-200 cm-1 higher than pob-TZVP with everything else in the input the same.
This is not a cimplaint, I understand that frequency outcome will depend on the calculation setup, e.g. functional and basis set, but why would pob-TZVP be so spectacularly bad at these Raman vibrations?
See attached, blue is 86111 old school basis set and orange is pob-TZVP basis set - I have much more data with pob-TZVP (unfortunately).
Experimental SO4 vibration is around 920 cm-1 in this particular mineral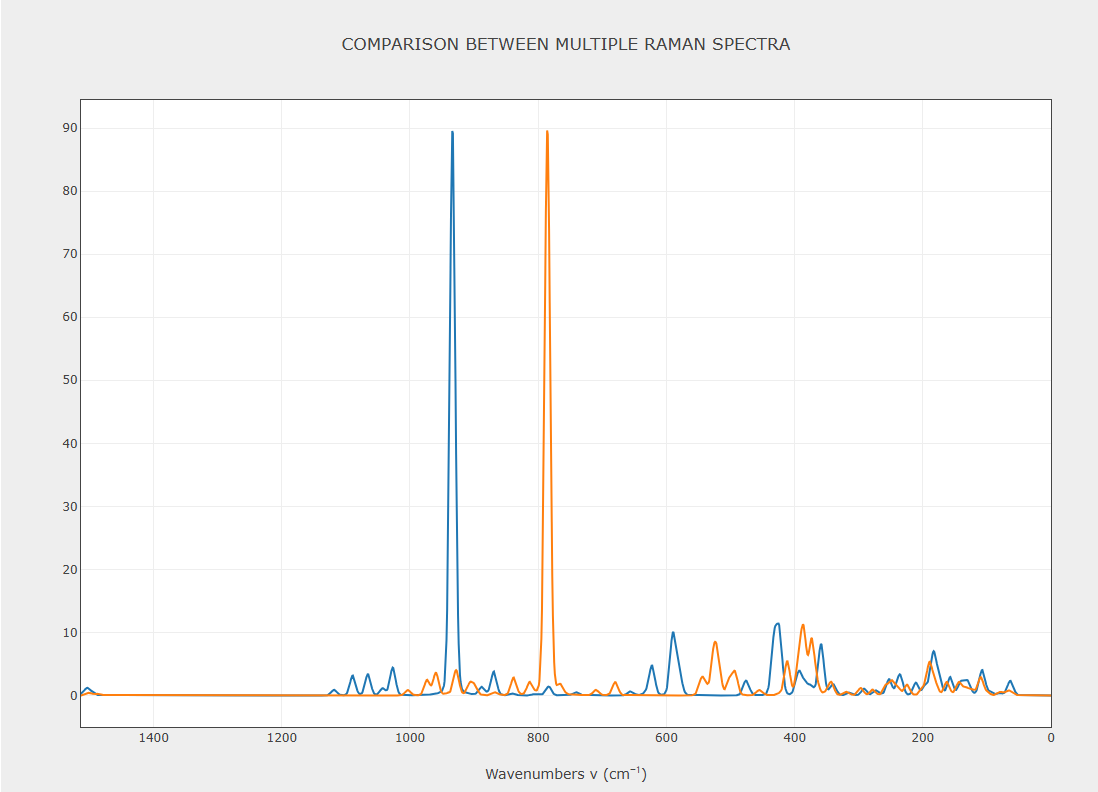
-
Hi,
Before sharing my general thoughts about POB versus "old school" basis sets, let me ask if you are using POB-TZVP or POB-TZVP-REV2 for your Sulfur-containing systems.
It was brought to my attention just a couple of days ago that the POB-TZVP-REV2 basis for Sulfur (S) is missing a whole d-type shell in the internal libraries in CRYSTAL and is thus not to be trusted, unless explicitly inserted in the input file as copy-pasted from the website!!!
-
-
59 S 0.802 -8.631 2.894
567 S
6.070E+04 5.470E-04 0.000E+00 0.000E+00
9.103E+03 4.230E-03 0.000E+00 0.000E+00
2.071E+03 2.175E-02 0.000E+00 0.000E+00
5.860E+02 8.510E-02 0.000E+00 0.000E+00
1.906E+02 2.480E-01 0.000E+00 0.000E+00
6.763E+01 4.670E-01 0.000E+00 0.000E+00
2.513E+01 3.643E-01 0.000E+00 0.000E+00
568 S
1.126E+02 2.167E-02 0.000E+00 0.000E+00
3.480E+01 9.360E-02 0.000E+00 0.000E+00
6.512E+00-2.607E-01 0.000E+00 0.000E+00
569 S
3.240E+00 1.284E+00 0.000E+00 0.000E+00
1.548E+00 6.604E-01 0.000E+00 0.000E+00
570 S
4.487E-01 1.000E+00 0.000E+00 0.000E+00
571 S
1.553E-01 1.000E+00 0.000E+00 0.000E+00
572- 574 P
5.644E+02 0.000E+00 2.480E-03 0.000E+00
1.334E+02 0.000E+00 1.968E-02 0.000E+00
4.247E+01 0.000E+00 8.998E-02 0.000E+00
1.562E+01 0.000E+00 2.571E-01 0.000E+00
6.109E+00 0.000E+00 4.352E-01 0.000E+00
575- 577 P
2.036E+00 0.000E+00 1.000E+00 0.000E+00
578- 580 P
4.338E-01 0.000E+00 1.000E+00 0.000E+00
581- 583 P
1.305E-01 0.000E+00 1.000E+00 0.000E+00 -
Yes, indeed, the d-type shell is missing. This is so unfortunate....
Worst case scenario is the only possible scenario it seems...
Although it is difficult to predict what effect this missing d orbitals will have on the computed frequencies, it may be significant.
Ok, so, you'll need to insert the basis set explicitly in the input for all elements in your system. That is, in the standard way, without the BASISSET keyword. For S, the complete pob-tzvp-rev2 basis is:
16 10
0 0 7 2.0 1.0
60700.928104 0.00054695944225
9102.6106854 0.00422972245570
2071.4166009 0.02174782415900
586.02476821 0.08510005358900
190.55395021 0.24799128459000
67.630384260 0.46703640406000
25.127306905 0.36434587550000
0 0 3 2.0 1.0
112.57463010 0.02167004024000
34.795554217 0.09360230176000
6.5115556215 -0.26068001422000
0 0 2 2.0 1.0
3.2399032261 1.28420894350000
1.5477160881 0.66036416584000
0 0 1 0.0 1.0
0.4487335200 1.00000000000000
0 0 1 0.0 1.0
0.1553457200 1.00000000000000
0 2 5 6.0 1.0
564.36716027 0.00247967963170
133.42624379 0.01967793025000
42.468271189 0.08998000825800
15.616527580 0.25705880575000
6.1093988469 0.43515167292000
0 2 1 4.0 1.0
2.0359436000 1.00000000000000
0 2 1 0.0 1.0
0.4337928300 1.00000000000000
0 2 1 0.0 1.0
0.1305009100 1.00000000000000
0 3 1 0.0 1.0
0.4107010100 1.0000000000000You can find the other basis sets and copy-paste them here
Although this is not my personal fault, I feel very sorry for this ugly bug.
-
Alessandro, the setback is temporary. What I really appreciate is that you gave CRYSTAL more life via user support than it ever had. I am very grateful for it

