help with substitutional defects
-
Alessandro,
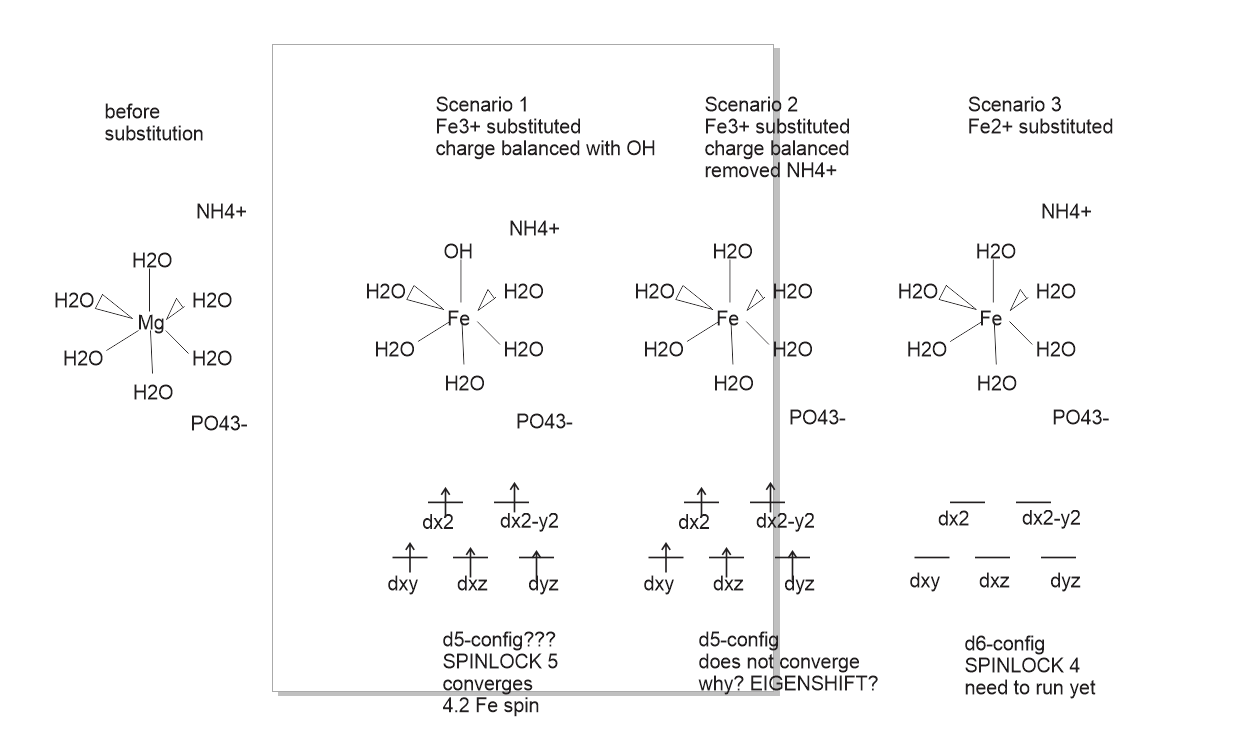
I am making a large 450 atom supercell of MgNH4PO4*6H2O to study Fe ion incorporation. We experimntally observe about 5% Mg atoms substituted with Fe - we do not know the oxidation state. So my supercell is big and I optimize this ionic solid with PBE-D3. My question is long and related on how to obtain required electronic state and converge in some instances. See below my diagram. I am exploring 3 different substitutional scenarios.
Scenario 1 is substituting Fe3+ and removing one proton to make it neutral. I am getting excellent SCF convergence, my guess is that this octahedral symmetry is broken somehow by removal of proton which helps SCF convergnce but crystal field with nearly 5 inpaired electrons is still preserved after the convergence with PBE-D3/pobDZVP.
Scenario 2 is an SCF problem. To charge balance it I remove NH4+. Immediately I get problems with SCF becoming conducting, I guess it does not want to be perfectly Oh symmetry and needs some distortion. I managed to converged it with PBE-D3/STO-3G in hopes that it will optimize to some solution which I can further optimize with PBE-D3/pobDZVP. But if I wanted to bias it with EIGENSHIFT and start with a large basus set, what would be my logic here? Robert Orlando used to very elegantly explain these things to me in the past.
Scenario 3 I haven't run yet. Seems like perfect Oh symmetry with the corresponding crystal field splitting and 4 unpaired electrons but my guess is it will run into the same problems as Scenario 2 and will need some biasing.
-
Hi Alessandro, working hard on this one. Scenario 3 seemed trivial (but did not converge). I assumed some JT distortion, so I used distortion along z axis argument and I shifted dxz down and assumed this is the double populated one. So far no luck in convergence
EIGSHIFT
5
1 8 1 -.5 .5
1 8 2 -.5 -.5
1 8 3 -.5 .5
1 8 4 -.5 .5
1 8 5 -.5 .5
-
-
Dear Jonas,
first of all, 450 atoms is impressive indeed!
Now moving on to a few comments/thoughts that might be of use to you.i) From your input file, it appears you still have a few symmetry operators in your simulation cell. Worth checking whether/if that applies to the atoms in the Fe-centred octahedra as that might force a particular solution to the electronic configurations.
ii) Given that your octahedra are not aligned with any particular crystallographic axis in your system, it is not always straightforward to extract the (d-)orbital occupations. You can have a look at the ROTREF keyword in the manual by which you can rotate the eigenvectors in the properties calculation to orient the frame along a principal axis.
iii) In Scenario 1 you are right - you removed 1 H atom, from which indeed the symmetry of the octahedron has been reduced/broken. You could test the case without "neutralization", to see whether the extra electron would stay on the site or delocalize...formation energies might be a good guide...?
iv) Scenario 2 is a bit more complicated. Given that you removed a whole NH4+ tetrahedron, I would expect significant structural distortions occurring in the surrounding of the defect. From there, a conducting state might be not so unrealistic - did you optimize the atomic positions and/or cell parameters following the inclusion of that defect? If so, could be worth seeing where the metallic states originate from (e.g., in the DOS or a charge density difference plot would be sufficient...don't forget SMEAR). The question is where is the extra electron from the anticipated Fe3+ ending if you remove the whole tetrahedron?
v) Finally, I guess it really depends on what you see experimentally and what would be the most representative simulation cell matching the measurements to be able to extract meaningful data for the oxidation state. What could be potentially useful is to take simple bulk structures for which you know the oxidation state of Fe (e.g., Fe2O3, Fe3O4, ...), analyse the corresponding charge densities (e.g., Mulliken) and have a reference state for comparing the charge on the Fe-ion in your Struvite structure.
Hope any of this is useful.
Cheers,
AleksAleksandar Živković, Scientific Assistant
Department of Earth and Environmental Sciences, LMU Munich, Germany
Crystal enthusiast
